
先天性短指畸形的发病机制、分类及治疗进展
2015-12-06 07:29:22杨茜综述王斌审校
组织工程与重建外科杂志 2015年6期
杨茜 综述 王斌 审校
先天性短指畸形的发病机制、分类及治疗进展
杨茜 综述 王斌 审校
先天性短指畸形(Brachydactyly,BD)是指由指(趾)骨或掌骨(跖骨)发育异常引起的一系列指(趾)缩短畸形。根据Bell的描述,短指畸形可分为A、B、C、D、E等五型。随着对短指畸形治疗的探索和研究的逐渐深入,BD又陆续被区分出若干亚型,其中,A3及D型最常见。研究发现,BD存在明显的家族遗传倾向,并与基因突变密切相关。本文综述了BD的国际分类、分子遗传学及治疗方案的研究进展,为BD的深入研究和治疗方案提供思路。
先天性短指畸形 家系研究 致病基因 治疗方案
先天性短指畸形(Brachydactyly,BD)是由指(趾)骨或(和)掌骨(跖骨)发育异常引起的一系列指(趾)缩短畸形。在Chang[1]对先天性上肢畸形的分类中,BD列属于“手板形成和分化障碍 (Failure in hand plate formation and differentiation)”。根据Hall在近年的国际疾病分类及基因相关性骨发育不良分类关于手部畸形的叙述中,BD列属于“肢体骨发育障碍”一组[2]。先天性短指畸形可表现为一种独立的疾病,也可表现为某些复杂的先天性畸形疾病的一部分。研究发现,BD存在明显的家族遗传倾向。近年来,在BD的家系研究中,发现了与短指畸形相关的一系列致病基因位点。
1 流行病学
随着人们生活水平以及认知水平的提高,短指畸形(BD)患者的就诊率较从前明显升高。但从目前各国的报道来看,BD仍属于罕见的先天性疾病,其发病率的统计差别较大。其中,A3及D型BD最常见[3]。但其发生率在不同族群间具有很大的差异性。A3型在不同人种中患病率为3.4%~21%,蒙古人种和美国印第安人中发病稍高。D型患病率为0.41%~ 4.0%,以色列阿拉伯人和日本人中患病率较高[4-6]。
2 国际分类
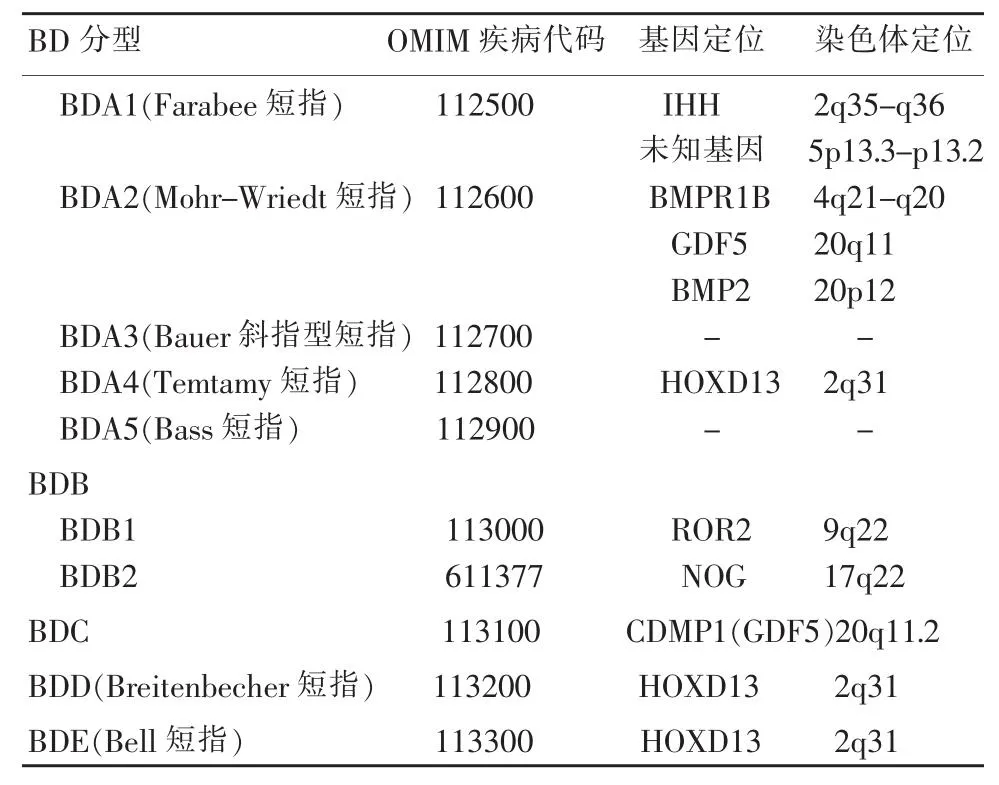
表1 BD分型及其基因定位Table 1 Classification and gene location of brachydactyly
根据Bell的描述,其将短指畸形分为A、B、C、D、E等五型 (表1)。由于对短指畸形治疗的探索和研究的逐渐深入,BD又陆续被区分出若干亚型(图1)。由于BD遗传异质性和新的分型不断被发现,因此根据表型诊断不易确定类型。我们主要依据Bell分类并结合在线人类孟德尔遗传目录(Online Mendelian Inheritance of Man,OMIM)[7]对单纯BD、部分伴发短指的综合症进行讨论。
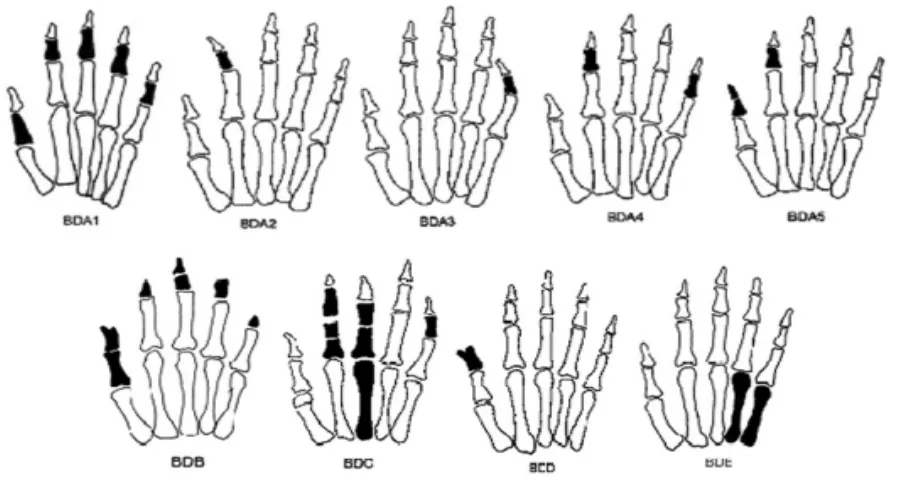
图1 短指畸形的分类示意图Fig.1 Classification diagram of brachydactyly
2.1 A型(Brachydactyly type A,BDA)
仅限于中节指骨短小畸形。根据受累指/趾的不同可分为1~5型。
2.1.1 A1型(Brachydactyly type A1,BDA1;MIM 112500)
2.1.1.1 临床及影像学特点
BDA1患者常以手指或足趾短小,影响功能及外形就诊,X线检查表现为典型的中节指/趾骨短小、退化,偶累及末节指/趾骨,伴有近节拇指/趾短小。其他一些临床表现还包括身材矮小和智力缺陷等。
根据缩短程度可将BDA1分为轻度和重度。轻度:中节指骨发育不全较轻,第2和5指/趾易受累,远节骨关节融合限于第5指/趾。重度:手指或足趾只有正常指/趾的一半,所有指/趾的中节指骨缺失或者严重发育不全,也可与远节指骨融合。指/趾骨短小畸形主要是由于骨骺缺失、骨干缩短以及骨骺发育过早终止所致,患者成年后身材短小[2-3]。
2.1.1.2 分子遗传学特点
BDA1是首例报道的人类孟德尔常染色体显性遗传性疾病。早在1908年,Farabee和Drinkwater首次发表了关于一个短指家系的研究,其后这个家系的表型被确定为Bell分型中的A1型短指,因此BDA1也称Farabee型短指/趾。近年来亦有关于BDA1的家系报道。目前已知的BDA1致病基因位点有至少两个:位于2q35-2q36的IHH(indian hedgehog gene)基因和位于5p13.2-p13.3之间区域的未知基因。其中,Ihh蛋白属于Hh(hedgehog)信号蛋白家族,是骨发育中心信号分子,控制软骨细胞分化、关节发育和骨形成[8-9]。柯新等[10]对一个中国山东BDA1型家系进行了报道,发现该家系为IHH基因发生了G298A(D100N)的错义突变所致,并将疾病相关单体型与Farabee-Drinkwater的疾病相关单体型进行了比较,表明该家系的G298A突变与白种人BDAl家系的IHH基因突变具有不同起源。目前,IHH基因在BDA1家系中已发现多个基因突变位点,而IHH基因的突变导致Ihh信号减弱是造成中间指(趾)节缩短或缺失的根本原因[11]。此外,Armour等[12]在一个有13名患者的3代家系中发现,在位于5p13.2-p13.3之间的D5S819到D5S1986未知基因区域与BDA1的发病相关。
2.1.2 A2型(Brachydactyly type A2,BDA2;MIM 112600)
2.1.2.1 临床及影像学特点
BDA2首次为Mohr和Wriedt所报道,常表现为第2指/趾典型的桡侧偏离。X线检查可见示指/第二个足趾中节指骨短小,余指/趾基本正常,受累中节指/趾骨通常呈长菱形或三角形且骨骺连续,故而出现第2指/趾远节向桡侧偏离[3]。该型BD特点与Jones[13]所描述的“Delta指骨”一致。
2.1.2.2 分子遗传学特点
目前已知的与BDA2有关的基因有BMPR1B、GDF5、BMP2等[6]。BMPR1B(bone morphogeneticprotein receptor 1B)基因位于4q21-q20,其产物是丝/苏氨酸激酶受体,BMPR1B的突变导致了对关节软骨生发的显性负性调控从而导致BDA2。此外,位于20q11的GDF5(Growth and differentiation factor 5)基因突变也可以导致出现BDA2的表型[14]。2009年,Dathe在一个无基因突变的BDA2家系中发现,位于20p12的BMP2(Bone morphogenetic protein 2)基因的下游重复与该家系的发病有关[15],Liu等[16]对一中国家系的研究同样支持了这一发现。
2.1.3 A3型(Brachydactyly type A3,BDA3;MIM112700)
2.1.3.1 临床及影像学特点
BDA3表现为仅第5指/趾中节指/趾骨短小畸形。第5指中节指骨短于第4指中节指骨的一半,X线显示有早期锥形骨骺。退化的中节指骨呈菱形或三角形,造成第5指向桡侧偏离。该表型较为常见,亦常在其他的疾病中出现。Williams在尼泊尔东部所做的1 357名3~20岁的幼儿-青年的流行病学调查显示,BDA3在该样本人群中的发病率为10.5%(男性12.3%,女性8.9%)[17]。事实上,该类型的BD是临床上造成斜指(Clinodactyly)畸形的常见原因[1]。
2.1.3.2 分子遗传学特点
BDA3表现为外显率不全的常染色体显性遗传,目前还未发现BDA3的致病基因或位点,国内外亦尚无关于BDA3的家系报道。
2.1.4 A4型(Brachydactyly type A4,BDA4;MIM 112800)
2.1.4.1 临床及影像学特点
BDA4在临床上较为罕见,该型BD属Bell分类中未分型,其首先为Temtamy和McKusick报道,发现于一个四代的家系中,因此又称“Temtamy短指”[3]。临床表现为第2和第5指中节指/趾骨短小,一些患者第4指/趾受累,且由于中节指/趾骨形状异常导致远节指骨桡侧偏离。下肢受累的患者通常第4趾中节趾骨缺失。有报道BDA4家系患者还伴先天性仰趾外翻足和畸形足。
2.1.4.2 分子遗传学特点
Reichenbach等[18]认为,BDA4符合常染色体显性遗传。Zhao等[19]在HOXD13基因(homeobox-containing gene)与人类肢体骨发育不全的相关性研究中,报道了一个涵盖了BDA4、BDD、BDE以及轻微并指畸形的庞大家系,并发现该家系为位于2q31上的HOXD13基因突变所致,提出以“HOXD13基因相关性肢体形态发育不全(HOXD13 limb morphopathies)”命名HOXD13基因突变所引起的一类肢体发育不全性疾病。目前尚无其他文献报道可区分BDA4、BDD、BDE的具有唯一相关性的致病基因。
2.1.5 A5型 (Brachydactyly type A5 with nail dysplasia;MIM 112900)
2.1.5.1 临床及影像学特点
BDA5属BDA中较严重的一种,临床表现为指/趾骨短缩伴指甲发育不全,X线检查可见中节指/趾骨缺失,拇指远节指/趾骨重复。Temtamy认为可将之归在BDB一类中[3]。
2.1.5.2 分子遗传学及家系研究
Bass[20]报道该型在家系中男-男传递,为常染色体显性遗传。目前对BDA5的分型尚存争议,亦无更多对BDA5家系及分子遗传学等的相关报道。
2.2 B型(Brachvdactyly type B,BDB)
BDB是最严重的一种BD,研究发现,BDB可由两种不同的致病基因突变而导致发病,故而在此将BDB分为B1及B2两个亚型。
2.2.1 B1型(Brachydactyly type B1,BDB1;MIM 113000)
2.2.1.1 临床及影像学特点
BDB1临床上类似截肢的表型,严重患者出现扁宽拇指,还可伴远节和/或中节指/趾骨末端分叉或双重远节指骨和并指。尺侧指/趾体受累一般较桡侧严重。X线表现为远节和/或中节指骨的短缩或发育不全,且常为对称性受累[3]。
2.2.1.2 分子遗传学特点
BDB1呈常染色体显性遗传模式,目前认为其致病基因为9q22上酪氨酸激酶受体ROR2(receptor tyrosine kinase-1ike orphan receptor 2)基因,ROR2属于“受体酪氨酸激酶样孤儿受体”家族,编码跨膜受体,其配体可能包括几种Wnts,还可以结合BMPRIB编码的受体[11]。近年来先后报道了多个ROR2基因的突变位点[21],其主要集中在外显子8和外显子9上。Huang等[22]对此进行了总结,并在临床上观察到一个中国家系,通过对该家系患者ROR2基因的外显子进行分析,发现该家系所有患者该基因于多核苷酸位点1396-1398处出现缺失(c.1396-1398delAA),这一新的突变造成了相关蛋白质的构象发生了变化。
2.2.2 B2型(Brachydactyly type B2,BDB2;MIM 611377)
2.2.2.1 临床及影像学特点
BDB2在临床表型上与BDB1类似,以远节指骨发育不全或缺失为特征,伴指骨关节融合,腕、跗骨融合,局部皮肤性并指。拇指短、拇指指甲发育不全或缺失,足部表型类似但略轻。
2.2.2.2 分子遗传学及家系研究
Lehmann在部分ROR2阴性的BDB合并偶发的近节指骨关节、腕、跗骨融合患者中(被认为是BDB2的表型),发现NOG(bone morphogenetic protein antagonist NOGGIN)基因的6个不同的突变,从而导致NOG与BMP结合能力的改变,并最终影响BMP信号通路的平衡[23]。NOG是一种糖基化的分泌蛋白,主要表达于神经组织和骨发育时期,在关节发育中起重要作用。Potti等[24]提出将包括BDB2在内的、有相同症状且与家族性NOG基因突变相关的疾病统一命名为NOGSSD(NOG-related symphalangismspectrum disorder)。
2.3 C型(Brachydactyly type C;MIM 113100)
2.3.1 临床及影像学特点
BDC患者临床上常表现为第4指相对受累最轻成为最长的指,而余指皆不同程度受累而出现短缩。患者足趾可正常或受累。X线典型表现为2、3、5指的中节指骨短缩,伴2和/或3指的指骨多节化,表现为明显“分节过多”,第1掌骨短小畸形。BDA与BDC临床上可出现相似的表现,其差异在于BDC涉及第一掌骨,而BDA没有;BDC中第4指/趾最长,很少受累,而BDA无此特点[3,25]。
2.3.2 分子遗传学特点
BDC呈常显、常隐等多种遗传方式,也有外显不全病例的报道,目前发现的唯一与BDC相关的基因是GDF5,又被称作 CDMP1(cartilage-derived morphogenetic protein-1)。GDF5属于转化生长因子TGF-β超家族,产物是分泌到细胞外的信号分子,通过上调Cbfal和X型胶原蛋白表达,促进肢芽细胞向软骨分化和促进骨形成[26]。当GDF5发生杂合移码或无义突变可造成BDC的表型。Uyguner等[27]观察到一个4代家系中9名不同程度发病的BDC患者的序列中存在一个新的插入缺失(c.803_827del25ins25),进而导致其编码GDF5蛋白的空间构象发生变化,最终影响蛋白功能。此外,GDF5基因另一位点的插入缺失突变与Du Pan综合征相关。
2.4 D型(Brachydactyly type D,BDD;MIM 113200)
2.4.1 临床及影像学特点
BDD又称为Breitenbecher短指,临床表现为拇指和第一足趾的远节指骨短而宽,呈“宽扁拇指”,其余指体不受累。X线检查可见拇指/趾远节指/趾骨宽扁,近节指/趾骨及掌/跖骨正常。有研究认为,BDD仅出现远节拇指/趾远节指/趾骨宽扁可能与骨骺线过早闭合有关[3]。该类型BD较为常见,发病率约在0.41%~4.0%之间。
2.4.2 分子遗传学及家系研究
BDD呈常染色体显性遗传,女性外显完全而男性外显不全,约3/4患者表现为双侧[15,28]。如前所述,BDD与BDE、BDA4都可由HOXD13基因突变引起。HOXD13基因是编码与肢体生长发育密切相关的同源异型结构域转录因子基因,Johnson等[28]在对 HOXD13基因的研究中发现错义突变与BDD、BDE的发生相关。Kuss在小鼠模型实验中发现,HOXD13的突变通过下调视黄酸水平,诱导指状组合型软骨形成,且与先天性并指/趾、多指/趾畸形显著相关[29]。这提示BD(至少是BDD、BDE)的发生与先天性并指/趾、多指/趾畸形的发生在分子水平上可能明显关联。
2.5 E型(Brachydactyly type E,BDE;MIM 113300)
2.5.1 临床及影像学特点
BDE以掌骨/跖骨短缩伴身材矮小、关节松弛为主要表现,Hertzog[30]认为BDE可分为3个亚型。E1型:限于第四掌/跖骨的短小畸形;E2型:多种掌骨或跖骨短缩畸形,合并第1、3指/趾远端指骨和第2、5指/趾中节指骨短缩;E3型:该型尚不确定,可能为多种掌骨短小但无指/趾骨畸形。由此可见,BDE表型存在明显异质性。小儿患者X线检查中可见骨发育不良及骨骺闭合。临床上BDE作为一种罕见类型,可单发亦可与一些复杂的先天畸形综合征伴发,如假性甲状旁腺功能减退症、高血压伴短指畸形(也称Bilginturan综合征)等等。
由此可见,BDE常难以判断其单发性或者为某种综合征的一部分,甚至在临床上易出现漏诊、误诊的情况。因此,Pereda等[31]在对BDE的研究中,结合儿科及内分泌科的工作,提出了一套标准诊断程序用以鉴别单纯性BDE和伴有掌骨短缩的综合征性疾病,如假性甲状旁腺功能减退症、高血压伴短指畸形等。
2.5.2 分子遗传学特点
BDE表现为常染色体显性遗传并外显不全。在单纯型BDE和BDE合并BDD患者中均发现HOXD13基因突变,而事实上临床中BDE的家系表型更为复杂。Pitt和Williams在其报道的一个四代的BDB和BDE的家系中提出了一类新的短指畸形,该家系患者有尺侧远节指骨发育不良合并一个或者多个掌骨短缩,Pitt将这类复杂表型的BD以该家系名字命名,为“BD Ballard型”[32]。Jensen等[33]报道了一个类似家系,但他们认为两个家系中的这种体征与E2亚型一致。
2.6 其他类型的BD
在临床上,对复杂型的BD描述亦不少见,早年有研究报道了一系列以趾骨、跖骨短缩症状为主的短指综合征[34-35],如单纯性跖骨短缩,近年来这类畸形更多地被报道在拇趾内翻、外翻的病例中[36]。
1927年,Kirner首先报道了小指桡侧倾斜,常双侧受累的先天畸形,命名为Kirner畸形,该类畸形相较于BDA3,除了小指中节指骨短缩外,还伴有远节指骨的发育不良和向桡侧弓状倾斜。近年来亦有对该畸形的报道,Satake等[37]提出将Kirner畸形按照弯曲程度分为骨骺线弯曲、骨干弯曲以及末节指骨弯曲3个亚型,临床上可据此结合患者的年龄对该畸形进行矫正。
其他有报道的特殊类型的BD见表2[38]。

表2 其他特殊类型的BDTable 2 Other specific types of brachydactyly

表3 与BD相关的综合征Table 3 BD related syndrome
2.7 部分与BD相关的综合征
2.7.1 Robinow综合征(胎儿面综合征)
1969年,Robinow等[39]首先报道了4例有明显畸形的患者,描述了一种罕见的以肢体中部短小、半椎体、特征性的面部畸形和生殖系统发育不足为主要特点的新的矮小综合征。该综合征的发病率非常低,约小于50万分之一。根据基因的不同将其分为两类:常染色体显性遗传性RS(Autosomal dominant form Robinow syndrome,DRS)和常染色体隐性遗传性RS(Autosomal recessive form Robinow syndrome,RRS)。两者比较,前者有更严重的肋骨和椎骨畸形[40]。目前认为,同BDB1一样,Robinow综合征可能与ROR2基因的删除(缺失)突变有关[41]。Wang等[42]认为,平面细胞极性信号(PCP signaling)的破坏可能是造成Robinow综合征和BDB1的基础。
2.7.2 du Pan综合征
du Pan综合症又称为腓骨发育不良合并复杂性短指/趾,特点为腓骨缺如,手足严重短指/趾,手指桡偏严重影响功能,是一类常染色体隐形遗传的先天性疾病。目前发现du Pan综合症与CDMP1基因(即GDF5基因)有关[43],Ahmad等[44]研究了一巴基斯坦家系,发现该家系du Pan综合症的发病是由于CDMP1基因leu441pro的替代突变造成的。
2.7.3 Temtamy轴前短指综合征
Temtamy轴前短指综合症是一类以双侧对称性的轴前短指、短趾,多节指合并扁平圆脸为主要特点的先天性综合征,常伴有牙齿发育异常、神经性耳聋、运动和智力以及生长发育迟缓等。呈常染色体隐形遗传,研究认为该疾病可能与CHSY1(Chondroitin Synthase 1)突变有关[45]。
2.7.4 其他
目前报道的与BD相关的综合征见表3。
3 治疗
就目前的医疗条件而言,尚无一种专门针对所有类型BD的标准术式。在能改善患者手足功能及外形的情况下可考虑行整形外科手术,但通常情况下该类手术也并非绝对必要。此外,术后的功能锻炼有助于手足功能恢复,也是重要的治疗环节[1]。
经典Bell分类有助于分子遗传学的研究,但该分类对BD的外科治疗意义不大,BD的治疗主要针对上肢进行,因此常借助Swanson关于上肢的先天畸形分类来作为治疗依据,其归类在第V组低度发育(Undergrowth)中[46]。
3.1 手术治疗
3.1.1 分指术
部分BD患者可合并并指畸形或手部亚单位结构不清,分指术是这类畸形必须最先考虑的手术。对于年幼患者而言,早期分指对促进指体长度具有意义,可能给予短缩的指体提供独立发育的基础。但是,目前分指术的适宜手术年龄尚无定论。
3.1.2 骨延长术(骨牵引术)
矫正短指畸形的主要手术是骨延长术,包括指骨延长及掌骨延长。目前该方法可平均延长10 mm(该数据为包含短指在内的、可行骨延长术的先天性上肢畸形的平均值),平均延长时间80 d左右,且7岁以下儿童骨延长效果较为理想[47]。但对于年纪尚小的幼儿患者需考虑其依从性。儿童骨延长术并发症较多,在治疗期间小儿可由于意外将延长螺丝拔出,或由于护理不良出现骨折、钉道感染等情况。此外,对于成功延长的指骨,亦有报道出现医源性血管神经损伤、旋转不良、捏力或握力下降、指体僵硬等并发症[48]。
在指骨尚无足够条件做骨牵引时,有时也可作局部软组织牵引,使局部形成足够长度的骨间隙[48],或称皮肤袋(skin pocket)[1],为接下来的游离植骨提供条件。
3.1.3 植骨术
对于严重的短指畸形,或严重的指骨发育不良形成融合性短指(Symbrachydactyly)的患者,或者骨延长术后软组织松弛、有骨移植条件的患者,可行趾骨、部分跖骨或者髂骨游离移植以补足手指的骨骼缺损,但移植效果取决于患者年龄、术后功能锻炼及手术水平等。植骨术在任何年龄都可进行,一般认为最适年龄不晚于2岁,但也有7岁患儿手术成功的报道。
此外,对于皮肤组织量不足的短指患者,还可以采用游离趾骨移植(Free phalangeal transfer)+扩大牵引成形术(Disdraction augmentation manoplasty)的方法。即患儿在进行趾骨移植术后再进行二期软组织牵引(一般不早于8岁),最后再进行游离骨移植。该方法的优点在于软组织伸缩性好,牵引时间短,使第二期手术可以在较短时间内完成,术后效果较单纯游离骨移植更好。
3.1.4 足趾移植术
严重的手发育不良、手指缺失,可行1个或2个足趾的带血管蒂移植,进行拇指及中指再造,或拇指、中指及环指的再造,最大限度重建对掌功能。目前认为该手术在3岁后进行效果较为理想[1]。一般情况下选择第二足趾趾骨进行移植,术前需仔细设计合适的供区肌腱、神经、血管以及合适的移植长度。Jones等[49]提出了详细的手术方案,为临床手术计划提供了参考。
3.1.5 其他手术选择
针对BDA2、BDA3患者或其他类型存在的指体偏斜、影响功能及外形者,可考虑行矫形手术或关节融合术。矫形术后配合功能锻炼,患者关节可以恢复部分活动度。但矫形手术对于骨骺的生长有很高的危险性,一旦骨骺破坏,将人为造成指骨的发育停止,因此手术多在患者骨骺接近闭合时或成年后进行[50]。
此外,对于分指术、植骨术或足趾移植后皮肤缺损可予以全厚皮片游离移植覆盖。
3.2 术后功能锻炼
患者术后的功能锻炼十分重要。患者自术后第2周可开始进行局部关节锻炼;拔出克氏针(第6周)后开始进行握力、捏力和对掌功能的训练,改善手指的灵活性,防止伤口及皮肤挛缩。
3.3 其他治疗
BD合并其他先天性畸形或者内分泌、心血管方面的疾病应进行相关治疗。
3.4 BD治疗的其他探索
3.4.1 肢体修复及肢芽生发的研究进展
随着再生技术的认识和发展,近年来也开始开展肢体修复再生及肢芽生发的研究,但目前研究尚停留在动物实验及分子遗传学研究阶段。如前所述,BD致病基因编码蛋白通过直接或间接的相互作用,形成了一个以GDF5/BMPRIB/ROR2为主的信号网络[12]。此外,胚胎干细胞(Embryonic stem cells ESCs)通过表达Lin28a,下调let-7水平,可以促进指体修复,为指体再生展示了良好前景[51]。
3.4.2 仿生手指、仿生机械手臂以及神经控制义肢的研究
对于个别严重病例,特别是手或足指/趾骨或掌/跖骨发育不良甚至缺失,手或者足功能严重丧失,单纯希望恢复外形的患者,可以根据患者手指正常生理长度和外形定制生物仿生手指(丙烯酸材料假指关节)[52]。另外,仿生机械手臂、神经控制义肢等方面的治疗仍在研究中,以脑电信息作为义肢信息源模式,尝试通过脑-机接口(Brain-Computer Interface,BCI)建立人与周围环境间信息交流与控制的新型通道,已经展开了一系列针对机械手臂的研究,更精细的功能重建也有望实现[53-54]。
4 总结
先天性短指畸形(BD)的研究已有百余年的历史,至今已基本建立起对该疾病的诊断、治疗以及发病机制研究的体系。BD是一类与基因突变密切相关的遗传病,各类BD的致病基因编码蛋白通过直接或间接的相互作用,形成一个以GDF5/BMPRIB/ROR2为主的信号网络。目前的研究也围绕这个网络不断深入,同时新的致病基因及发病机制亦不断被发现。治疗方面,除了优化传统的手术方法之外,再生医学为肢体修复和肢芽生发展示了良好前景,而医工结合的相关的仿生手指、仿生机械手臂以及神经控制义肢的研究,为严重短指畸形的治疗提供了不同的解决方案。总之,随着BD分子水平研究的开展和再生医学的快速发展,先天性短指畸形的研究有望在将来为其临床治疗带来突破。
[1] Chang J.Plastic Surgery Volume 6 Hand and Upper Extremity [M],3rd edition.Stanford:Elsevier Saunders,2013:550-641.
[2] Hall CM.International nosology and classification of constitutional disorders of bone[J].Am J Med Genet,2002,113(1):65-77.
[3] Temtamy S,Aglan MS.Brachydactyly[J].Orphanet J Rare Dis, 2008,3(1):1-16.
[4] Mundlos S.The brachydactylies:a molecular disease family[J]. Clin Genet,2009,76(2):123-136.
[5] Temtamy S,Mckusick VA.The genetics of hand malformations [J].Birth Defects Orig Artic Ser,1978,14(3):16-19.
[6] 虢毅,梁卉,邓昊.短指/趾的分子遗传学研究进展[J].遗传,2012, 34(12):1522-1528.
[7] Amberger J,Bocchini CA,Scott AF,et al.McKusick's online mendelian inheritancein man(OMIM)[J].Nucleic Acids Res, 2009,37(Suppl 1):D793-D796.
[8] 周坚.IHH和FGF8在脊椎动物肢翼远端骨骼元件发育中的功能研究[D].上海:上海交通大学,2008.
[9] Gao B,Hu JX,Stricker S,et al.A mutation in Ihh that causes digit abnormalities alters its signaling capacity and range[J]. Nature,2009,458(7242):1196-1200.
[10] 柯新,董爱玲,刘奇迹.一个短指家系临床特征调查及其致病基因的定位[J].中华医学遗传学杂志,2009,26(3):267-271.
[11] 杨威.短指(趾)和缺指(趾)畸形的分子遗传学研究[D].北京:中国协和医科大学,2008.
[12] Armour CM,MeCready ME,Baig A,et al.A novel locus for brachydactyly type A1 on chromosome 5p13.3-p13.2[J].J Med Genet,2002,39(l):186-188.
[13] Jones GB.Delta phalanx[J].J Bone Joint Surg,1964,45:1704.
[14] Lehmann K,Seemann P,Stickler S,et al.Mutations in bone morphogenetic proteinreceptor 1B causebrachydactyly type A2 [J].Proc Natl Acad Sci USA,2003,100(21):12277-12282.
[15] Dathe K,Kjaer KW,Brehm A,et al.Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2[J].Am J Hum Genet,2009,84(4):483-492.
[16] Liu X,Gao L,Zhao A,et al.Identification of duplication downstream of BMP2 in a Chinese family with brachydactyly type A2 (BDA2)[J].PLoS ONE,2014,9(4):e94201.
[17] Williams KD,Blangero J,Cottom CR,et al.Heritability of brachydactyly type A3 in children,adolescents,and young adults from an endogamous population in eastern Nepal[J].Hum Biol, 2007,79(6):609-622.
[18] Reichenbach H,Hormann D,Theile H.Brachydactyly type A4 (brachymesophalangia II and V,Temtamy type):A rare type of brachydactyly[J].Kinderarztl Prax,1993,61(2):59-62.
[19] Zhao X,Sun M,Zhao J,et al.Mutationsin HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome [J].Am J Hum Genet,2007,80(2):361-371.
[20] Bass HN.Familial absence of middle phalanges with naildysplasia: a new syndrome[J].Pediatrics,1968,42(2):318-323.
[21] Schwabe GC,Tinschert S,Buschow C,et al.Distinct mutations in the receptor tyrosine kinase geneROR2 cause brachydactyly type B[J].Am J Hum Genet,2000,67(4):822-831.
[22] Huang D,Jiang S,Zhang Y.A new mutation in the gene ROR2 causes brachydactyly type B[J].Gene,2014,15:547(1):106-110.
[23] Lehmann K,Seemann P,Silan F,et al.A new subtype of brachydactyly type B caused by point mutations in the bone morphogenetic protein antagonist NOGGIN[J].Am J Hum Genet,2007,81(2): 388-396.
[24] Potti TA,Petty EM,Lesperance MM.Comprehensive review of reported heritable Noggin-associated syndromes and proposed clinical utility of one broadly inclusive diagnostic term:NOG-related-symphalangism spectrum disorder(NOG-SSD)[J].Human Mutation,2011,32(8):877-886.
[25] Robin NH,Gunay-Aygun M,Polinkovsky A,et al.Clinical and locus heterogeneity in brachydactyly type C[J].Am J Med Genet, 1997,68(3):369-377.
[26] 吕学敏,邓廉夫,杨庆铭,等.生长分化因子—5影响肢芽细胞分化的机制[J].中华骨科杂志,2005,25(6):368-371.
[27] Uyguner ZO,Kocaoglu M,Toksoy G,et al.Novel indel mutation in the GDF5 gene is associated with brachydactyly type C in a four-generation Turkish family[J].Mol Syndromol,2014,5(2):81-86.
[28] Johnson D,Kan SH,Oldridge M,et al.Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E[J].Am J Hum Genet,2003,72(4):984-997.
[29] Kuss P,Villavicencio-Lorini P,Witte F,et al.Mutant Hoxd13 induces extra digits in a mouse model of synpolydactyly directly and by decreasing retinoic acid synthesis[J].J ClinInvest,2009, 119(1):146-156.
[30] Hertzog KP.Brachydactyly and pseudo-pseudohypoparathyroidism [J].Acta Genet MedGemellol,1968,17(3):428-438.
[31] Pereda A,Garin I,Garcia-Barcina M,et al.Brachydactyly E: isolated or as a feature of a syndrome[J].Orphanet J Rare Dis, 2013,12(8):141.
[32] Pitt P,Williams I.A new brachydactyly syndrome with similarities to Julia Bell types B and E[J].J Med Genet,1985,22(3):202-204.
[33] Jensen K,Hoo JJ.Is brachydactyly type Ballard a variant of brachydactyly type E[J]?Am J Med Genet A,2004,129(1):95-97.
[34] Ray AK,Haldane JB.The genetics of a common Indian digitalabnormality[J].Proc Nat Acad Sci,1965,53(5):1050-1053.
[35] Sugarman GI,Hager D,Kulik WJ.A new syndrome of brachydactyly of the hands and feet with duplication of the first toes[J].Birth Defects Orig Art Ser,1974,10(5):1-8.
[36] Froehlich V,Wuenschel M.A rare combination of brachymetatarsiaand congenital hallux varus:case report and review of the literature [J].J Am Podiatr Med Assoc,2014,104(1):85-92.
[37] Satake H,Ogino T,Eto J,et al.Radiographic features of Kirner's deformity[J].Congenit Anom,2013,53(2):78-82.
[38] Mckusick V.Mendelian inheritance of man.A catalogue of human gene and genetic disorders[M].Baltimore:JohnsHopkins University Press,1994.
[39] Robinow M,Silverman FN,Smith HD.A newly recognized dwarfing syndrome[J].Am J Dis Child,1969,117(6):645-651.
[40] 唐晓军,尹琳,张智勇.Robinow综合征[J].中华整形外科杂志, 2010,26(5):391-393.
[41] Stickler S,Verhey van Wijk N,Witte F,et al.Cloning and expression pattern of chicken Ror2 and functional characterization of truncating mutations in Brachydactyly type B and Robinow syndrome[J]. Dev Dyn,2006,235(12):3456-3465.
[42] Wang B,Sinha T,Jiao K,et al.Disruption of PCP signaling causes limbmorphogenesis and skeletal defects and may under lie Robinow syndrome and brachydactyly type B[J].Hum Mol Genet,2011,20 (2):271-285.
[43] Faiyaz-Ul-Haque M,Ahmad W.Mutation in the cartilage-derived morphogenetic protein-1(CDMP1)gene ina kindred affected with fibular hypoplasia and complex brachydactyly(DuPansyndrome) [J].Clin Genet,2002,61(6):454-458.
[44] Ahmad M,Abbas H,Wahab A,et al.Fibular hypoplasia and complex brachydactyly(Du Pan syndrome)in an inbred Pakistani kindred[J].Am J Med Genet,1990,36(3):292-296.
[45] Li Y,Laue K,Temtamy S,et al.Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in Chondroitin Synthase 1,a potential target of BMP signaling[J].Am J Hum Genet,2010(87):757-767.
[46] Swanson A.A classification for congenital limb malformations[J]. J Hand Surg Am,1976,1(1):8-22.
[47] Matsuno T,Ishida O,Sunagawa T,et al.Bone lengthening for congenital differences of the hands and digits in children[J].J Hand Surg Am,2004,29(4):712-719.
[48] Nguyen ML,Jones NF.Salvage reconstruction of failed pollicization by distraction lengthening[J].Hand,2011,6(3):324-328.
[49] Jones NF,Kaplan J.Indications for microsurgical reconstruction of congenital hand anomalies by toe-to-hand transfers[J].Hand, 2013,8(4):367-374.
[50] 刘旭东,范存义,曾炳芳.先天性A2型短指症的矫形手术治疗[J].中华手外科杂志,2011,27(3):136-137.
[51] Shyh-Chang N,Zhu H,Yvanka de SoysaT,et al.Lin28 enhances tissue repair by reprogramming cellular metabolism[J].Cell, 2013,155(4):778-792.
[52] Rajeev KRK,Vinod B,Bharathi M,et al.Acrylic finger prosthesis: a case report[J].J Clin Diagnostic Res,2014,8(8):7-8.
[53] 李耀楠,张小栋,王云霞.脑一机接口驱动经义肢手系统的研究[J].中国医疗设备,2011,4(2):1674-1633.
[54] Amsuess S,Goebel P,Graimann B,et al.A multi-class proportional myocontrol algorithm for upper limb prosthesis control:validation in real-life scenarios on amputees[J].IEEE Trans Neural Syst Rehabil Eng,2015,23(5):827-836.
Etiology,Classification and Treatment of Brachydactyly
YANG Xi,WANG Bin.Department of Plastic and Reconstructive Surgery,Shanghai Ninth People's Hospital,Shanghai Jiaotong University School of Medicine,Shanghai 200011,China.Corresponding author:WANG Bin(E-mail:wangbin1766@163.com).
【Summary】 Brachydactyly(BD)is a congenital deformity that refers to shortening of the hands/feet due to small or missing metacarpals/metatarsals and/or phalanges characterized by bone dysostosis.According to Bell's classification,BD can be divided into to five categories:A to E.Recent years,new subtypes were advocated.Among these,BDA3 and BDD may be the most common subtypes.The deformity was close related to familial inheritance and gene mutations.In this review,the advancements in etiology,classification and treatments of brachydactyly were summarized.
Brachydactyly; Family study;Pathogenic gene;Treatment
R622
B
1673-0364(2015)06-0389-07
10.3969/j.issn.1673-0364.2015.06.012
2015年9月10日;
2015年10月8日)
上海市重中之重临床医学中心。
200011 上海市 上海交通大学医学院附属第九人民医院整复外科。
王斌(E-mail:wangbin1766@163.com)。
猜你喜欢
实用手外科杂志(2022年2期)2022-08-31 09:47:48
体育科研(2021年3期)2021-05-29 08:44:12
畅谈(2018年19期)2018-01-22 08:36:02
外科研究与新技术(2016年2期)2016-12-22 02:29:34
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
实用手外科杂志(2015年2期)2015-08-28 09:50:40
实用手外科杂志(2015年4期)2015-08-27 01:54:08
实用手外科杂志(2015年1期)2015-08-27 01:52:08
