
电沉积方式对制备氧化锌薄膜的影响
2015-12-05 09:18:14彭友舜王晓娟张丽茜秦秀娟
电镀与精饰 2015年9期
彭友舜, 王晓娟, 张丽茜, 秦秀娟
(1.河北科技师范学院 化学工程学院,河北秦皇岛 066004;2.燕山大学环境与化学工程学院,河北秦皇岛 066004)
引 言
ZnO是一种典型的Ⅱ~Ⅵ族直接带隙宽禁带半导体,室温下带宽为 3.37eV,激子结合能高达60meV,室温下能观察到ZnO的紫外发射。ZnO半导体材料具有较宽的带隙宽度、较高的化学稳定性、无毒等优点,使其在光电、压电、光催化及太阳能电池等领域有广阔的应用前景,因而成为当前材料研究的热点。
制备ZnO薄膜常用的方法有磁控溅射法[1-2]、溶胶-凝胶(Sol-Gel)法[3]、化学气相沉积(CVD)法[4]和脉冲激光沉积(PLD)法[5]等,上述方法可以制备出质量较高的ZnO薄膜,但工艺复杂[6],反应温度高[7],反应条件不易控制,所以反应条件温和、实验装置简单及易于控制样品形貌的电沉积法受到人们的关注。
电沉积方式分为恒电流与恒电位两大类,两种沉积方式都能得到理想的纳米氧化锌材料[8-10]。但对两种沉积方式所获得的薄膜的性能差异少有关注。氧化物沉积量只有在沉积初期与理论值较为接近,以后随时间推移,二者偏差越来越大。由于恒电位与恒电流过程中,电流与电压都在变化,所以在这个意义上电位与电流的区别很难更深刻的解释两种沉积方式的特点。本文采用两种沉积方式制备了纳米氧化锌薄膜,在光学性能、表面形貌和内部缺陷等方面分析了沉积方式对纳米ZnO薄膜性能及缺陷的影响,进一步丰富了利用电沉积制备纳米薄膜的内容,对不同领域应用纳米ZnO薄膜的可控生长有一定的指导意义。
1 实验
1.1 试剂与仪器
实验所用的试剂Zn(NO3)2·6H2O(分析纯)、无水乙醇、硝酸、丙酮(均为天津化学试剂厂)。去离子水采用实验室自制的二次去离子水。基底材料是ITO(In2O3∶Sn)导电玻璃(表面方阻为10~15Ω)。
1.2 氧化锌薄膜的制备
氧化锌薄膜的制备采用传统的三电极体系,饱和甘汞电极(SCE)为参比电极,铂电极为辅助电极,ITO导电玻璃作为工作电极(15mm×15mm×3mm);工作电极与辅助电极平行放置,间距2cm;电解液由0.07mol/L硝酸锌水溶液组成,反应前溶液pH为5.5(稀硝酸标定),实验在65℃的恒温水浴环境下进行;沉积t为10min。反应过程中使用磁力搅拌器搅拌,转速保持300r/min。反应完成后将样品取出,用去离子水多次冲洗,室温条件下自然晾干,得到所制备的样品。
1.3 样品的检测
本文对样品的X-射线衍射谱测量采用D-max-2500/PC型X-射线衍射仪(日本理学株式会社公司),Cu靶为入射光源,扫描速率4°/min;样品表面形貌分析使用MultiMode 8 SPM型原子力显微镜(布鲁克 AXS公司);样品在室温下的光致发光(PL)性能通过FL3-11型荧光光谱仪测量,激发光源为Xe激光器,激发波长为325nm;使用WFZ-26A型紫外-可见分光光度计在室温下测量样品的UVVis吸收光谱,测量范围为200~800nm。
2 结果与讨论
2.1 沉积过程的研究
由硝酸锌电沉积制备氧化锌的反应过程:
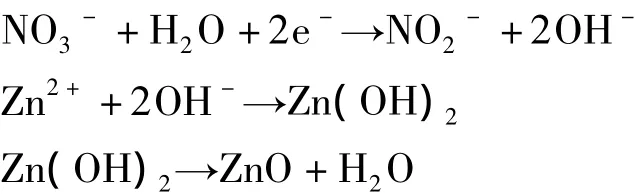
在电解过程中,阴极同时发生了如下副反应:
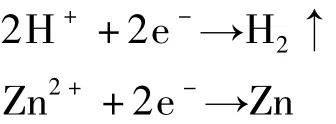
由于在电解过程中有析氢和析锌副反应发生,会影响制备薄膜的质量,因此沉积电位或电流的大小往往是电沉积法所要解决的首要问题。
图1为阴极极化曲线。从图1中可知,在电位达到-0.5V之前电流密度几乎为零;随着电位逐步增加,渐渐有微小的电流出现,发生了析氢副反应;电位继续增加,电流逐渐增大,溶液发生NO-3的还原反应,逐步生成ZnO;当电位达到-1.0V时电流迅速增大,电解以显著速率进行,同时电位的继续增加还可能引发金属锌的析出。为了确保主反应的快速进行,抑制副反应(析氢和析锌),沉积电位的选择在-0.7~-1.0V之间,选择沉积电位为-1.0V,根据其相对应的电流-时间曲线选取对应稳定的电流值 -0.13A/dm2,作为研究的比较对象。
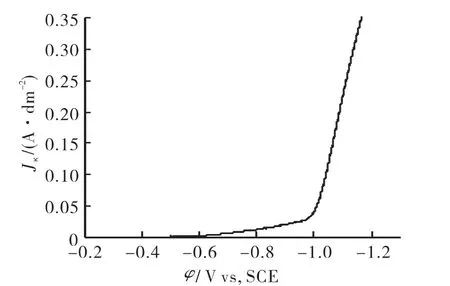
图1 Zn(NO3)2溶液的阴极极化曲线
通过测量、分析恒电位沉积过程中的电流密度-时间曲线和恒电流沉积过程中的电位-时间曲线来推论氧化锌的沉积过程,比较两种沉积方式在机理上的不同。如图2所示。
在恒电位沉积过程中电流在反应起始阶段有一个突然减小的过程见图2(a),这与氧化锌种子层的生成有关[11];表明沉积电位越大这一过程进行的越迅速,即种子层生成越快。图2(b),恒电流沉积的起始阶段由于要生成氧化锌种子层,阴极电位有一个突然增大(变负)的过程,最大电位值接近-1.3V,相对于恒电位过程的 -1.0V,种子层的生成更加迅速、致密。
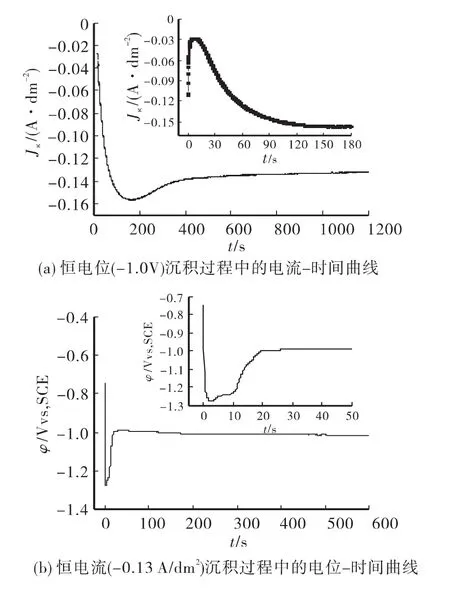
图2 电沉积过程中的电位-时间曲线
图3为两种电沉积方法制得样品的UV-Vis吸收光谱图。图中显示实验中恒电流沉积的ZnO薄膜的透过性要优于恒电位沉积的样品,恒电流沉积的薄膜在480nm时透射比就达到80%。图3中的插图为ZnO半导体材料的Tauc曲线,也证实了恒电流沉积的薄膜的透光性好于恒电位沉积的样品。这是因为恒电流沉积的初始阶段,为了保持恒定的电流值,阴极极化电位迅速增大,使得在很短的时间内快速生成大量ZnO种子颗粒,其过程见图4。沉积电流越大这一过程用时越短,生成的种子层也越均匀致密[12],有利 ZnO 的生长,解释了文献[13]报道中在沉积之前使用高电位短时间沉积种子层的原因。
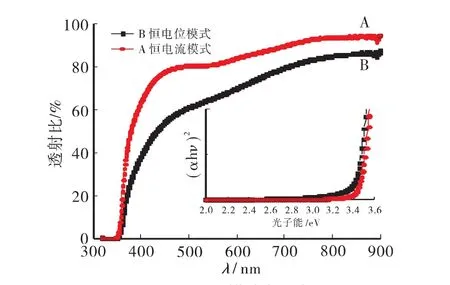
图3 不同沉积模式制得样品透射光谱和(αhν)2vs hν图

图4 使用不同沉积模式电沉积ZnO薄膜反应历程示意图
当沉积电流密度过大,反应起始阶段迅速增大的阴极电位值有可能达到Zn2+的析出电位(-1.05V),析锌对薄膜的性能会产生不利的影响,因此当沉积电流密度过大时,非但不能提高结晶质量,反而会影响薄膜的性能。从这点上讲,恒电流法的电流选择范围较窄。
2.2 结构与形貌分析
图5为硝酸锌水溶液采用不同沉积方式制备ZnO薄膜的X-射线衍射谱图(XRD)。样品A的X-射线衍射峰晶面(100)、(002)、(101)、(102)、(110)、(103)及(112),分别对应的 2θ=31.642°、34.298°、36.118°、47.408°、56.427°、62.702°及67.757°,衍射峰峰位与标准 ZnO(PDF-36-1451)的衍射峰相吻合,样品B的衍射峰位也与其基本对应。证明制得的样品为多晶六角纤锌矿(hexagonal wurtzite)结构的ZnO薄膜。图5中未发现杂质及基底的衍射峰,说明制备的ZnO薄膜结晶质量良好、纯度高及基底覆盖性好。不同沉积方式的XRD谱图无明显区别。制得的薄膜没有文献报道[14]中的(002)择优取向。
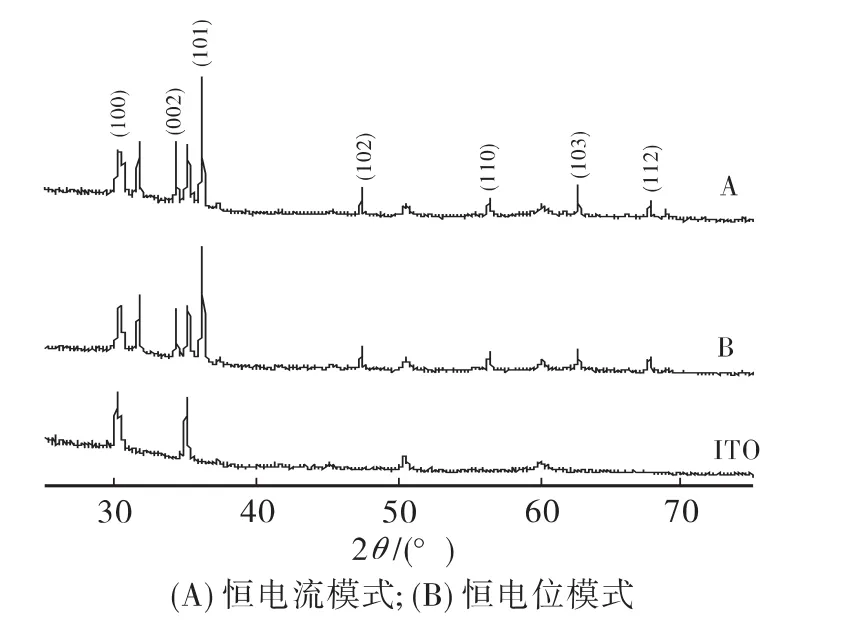
图5 不同沉积模式制得样品的XRD谱图
图6、图7为两种电沉积方法制得的AFM照片。由图6、图7可知,恒电位沉积的氧化锌薄膜呈类球型,颗粒尺寸较均匀;恒电流沉积的氧化锌薄膜为片状,呈纳米壁式堆积,彼此接触的地方生长在一起,形成与恒电位沉积相同的小山丘状。这与之前分析的反应历程相吻合,由于恒电流反应起始阶段快速生成致密的种子层,后续氧化锌薄膜的生长得以均匀、平稳的进行。相比于恒电位沉积,恒电流沉积制得的薄膜表面比较平整(表1)。
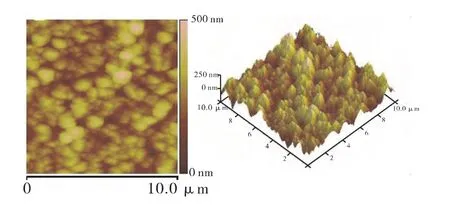
图6 恒电位模式制备的ZnO薄膜AFM图

图7 恒电流模式制备的ZnO薄膜AFM图
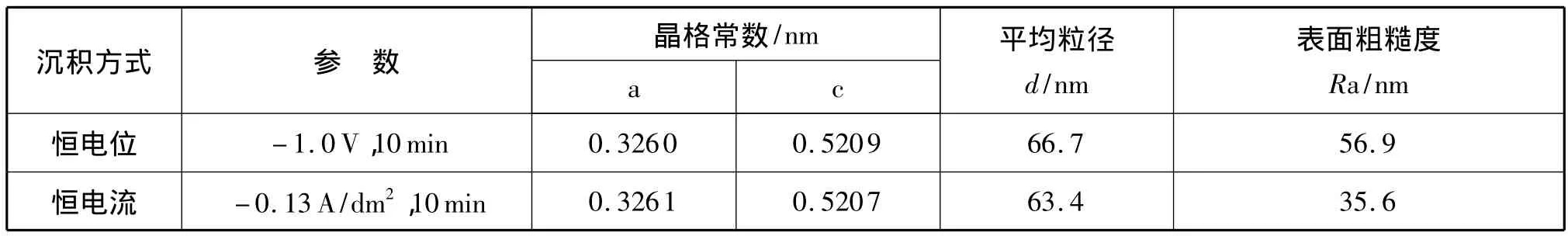
表1 不同沉积方式制得ZnO薄膜的特性参数
2.3 荧光-可见光性能分析
两种沉积方式制备的氧化锌薄膜在397nm附近都有一个紫外近带边发射峰(图8),被认为是属于氧化锌的激子发射。恒电位沉积得到的薄膜在509nm处有一缺陷发射峰,与文献[15]报道的510nm附近的绿色发光峰相吻合。恒电流法所得的薄膜缺陷发射峰成一馒头状,位于540nm处。计算得510nm缺陷发射峰对应能量为 2.44eV,而540nm缺陷发射峰对应能量为2.30eV。根据 Xu等[16]利用全势能线性多重轨道(FP-LMTO)方法计算的ZnO中的5种本征缺陷和复合缺陷:氧空位(VO)、锌空位(VZn)、氧填隙(Oi)、锌填隙(Zni)和氧错位(OZn),从导带底到氧替位缺陷能级的能量差为2.39eV,与实验中观测到的恒电位沉积制备的样品缺陷发射峰能量基本一致;氧填隙(Oi)能级与导带底的能级间隔(2.29eV)近似满足恒电流沉积制备样品的缺陷发射峰。推断,恒电流沉积与恒电位沉积制备的氧化锌存在着不一样的缺陷,前者主要集中在黄色发光峰,对应于氧填隙缺陷,而后者主要集中在绿色发光峰,为氧错位缺陷。
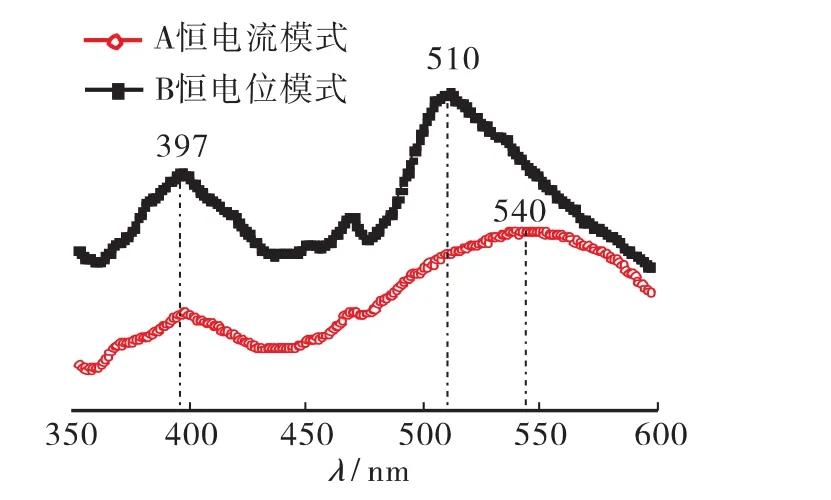
图8 不同沉积模式制备的ZnO薄膜PL光谱图
3 结论
通过比较分析恒电位与恒电流两种沉积方式在制备氧化锌沉积过程的差异,探讨了不同沉积方式制备ZnO薄膜的机理。与恒电位沉积方式相比,恒电流沉积的初始阶段有利于种子层致密、快速的生成,而稳定的大电流又有利于细化结晶颗粒,对薄膜的透过性能有较大改善作用;考虑到沉积过程中的副反应(析氢、析锌),恒电流模式下电流的选择范围要小于恒电位模式下电位的选择范围;不同沉积方式制备的氧化锌薄膜存在不同的缺陷,形成原因可能与反应进程的快慢有关。
[1] 夏至美,刘竹林.氧化锌掺杂改性研究进展[J].材料导报,2014,21(3):47-52.
[2] Abdelouahab Gahtar,Achour Rahal,Boubaker Benhaoua,et al.A comparative study on structural and optical properties of ZnO and Al-doped ZnO thin films obtained by ultrasonic spray method using different solvents[J].International Journal for Light and Electron Optics,2014,125(14):3674-3678.
[3] Prakash Chand,Anurag Gaur,Ashavani Kumar,et al.Structural,morphological and optical study of Li doped ZnO thin films on Si(100)substrate deposited by pulsed laser deposition[J].Ceramics International,2014,40(8):11915-11923.
[4] Zunke I,Wolf S,Heft A,et al.Structural properties of zinc oxide deposited using atmospheric pressure combustion chemical vapour deposition[J].Journal of Applied Physics,2014,565(1):45-53.
[5] Hye-Ji Jeon,Seul-Gi Lee,Kyung-Sik Shin,et al.Growth behaviors and film properties of zinc oxide grown by atmospheric mist chemical vapor deposition[J].Journal of Alloys and Compounds,2014,614(1):244-248
[6] Ha-Rim An,Hyo-Jin Ahn,Jeong-Woo Park,et al.Ga-doped ZnO films grown by atmospheric-pressure chemical-vapor deposition[J].Ceramics International,2015,2(A):2253-2259.
[7] Prepelita P,Medianu R,Sbarcea B,et al.Characterizations of arsenic-doped zinc oxide films produced by atmospheric metal-organic chemical vapor deposition[J].Applied Surface Science,2013,277(1):1-6.
[8] Fanni L,Aebersold B A,Alexander D T L,et al.c-texture versus a-texture low pressure metalorganic chemical vapor deposition ZnO films:Lower resistivity despite smaller grain size[J].Thin Solid Films 2014,565(1):1-6.
[9] Ding L,Fanni L,Messerschmidt D,et al.Tailoring the surface morphology of zinc oxide films for high-performance micromorph solar cells[J].Solar Energy Materials and Solar Cells,2014,128(1):378-385.
[10] 胡友娇,杨冬梅,杨昌柱,等.ZnO薄膜光电催化降解乙酰甲胺磷[J].环境科学与技术,2012,1(3):335-340.
[11] 刘永利,刘欢,李蔚.衬底温度对ZnO薄膜生长过程和微结构的影响[J].物理化学学报,2013,29(3):631-638.
[12] Kamila Zarebska,Maciej Kwiatkowski,Marianna Gniadek,et al.Electrodeposition of Zn(OH)2,ZnO thin films and nanosheet-like Zn seed layers and influence of their morphology on the growth of ZnO nanorods[J].Electrochimica Acta,2013,98(30):255-262.
[13] 张利静,方华靖,严清峰.种子层诱导溶胶-凝胶法Li,Mg掺杂ZnO薄膜的结构及光电性能研究[J].人工晶体学报,2013,42(7):1272-1277.
[14] 尹玉刚,沈鸿烈,楼晓波.溶胶-凝胶法生长(002)高度择优取向的 ZnO:Al薄膜[J].功能材料,2008,39(7):1122-1125.
[15] 袁艳红,侯洵,高恒.超声处理对ZnO薄膜光致发光特性的影响[J].物理学报,2006,5(1):446-449.
[16] Xu P S,Sun Y M,Shi C S,et al.The electronic struc-ture and spectral properties of Zno and its defects[J].Nuclear Instruments and Methods in Physics Research B,2003,199:286-290.
猜你喜欢
建材发展导向(2023年5期)2023-03-15 03:30:10
食品安全导刊(2021年20期)2021-11-28 00:56:56
光源与照明(2019年4期)2019-05-20 09:18:18
电线电缆(2017年2期)2017-07-25 09:13:35
电镀与环保(2016年2期)2017-01-20 08:15:26
现代工业经济和信息化(2016年12期)2016-05-17 05:37:52
现代工业经济和信息化(2016年1期)2016-05-17 05:33:21
物理化学学报(2015年7期)2015-12-30 12:13:16
电源技术(2015年12期)2015-08-21 08:58:58
中国药业(2014年21期)2014-05-26 08:56:34

- 电镀与精饰的其它文章
- 《电镀与精饰》2016年期刊征订启事