
钴(Ⅱ)催化CpCoS2C2B10H10分子内B(3)/B(6)位和Cp配体偶联
2015-11-30 08:41:19叶红德徐宝华胡久荣燕红
无机化学学报 2015年7期
叶红德 徐宝华 胡久荣 燕红*,
钴(Ⅱ)催化CpCoS2C2B10H10分子内B(3)/B(6)位和Cp配体偶联
叶红德*,1,2徐宝华1胡久荣2燕红*,1
(1南京大学化学化工学院,配位化学国家重点实验室,南京210093) (2上饶师范学院化学化工学院,上饶334001)
半夹芯16e化合物CpCoS2C2B10H10(Cp:cyclopentadienyl)(1)与HC≡CCO2Me在2-甲基二硫代丙酸存在下反应生成化合物{(C5H4CoS2C2B9H9)(CH=CHCO2Me)(Me2C=CS2H)}(2)和(Me2C=CS2H)3Co(3)。在化合物2中,原料化合物1中的一个S-Co键断裂,该S原子与一分子HC≡CCO2Me末端炔基碳原子连接。Co原子与2-甲基二硫代丙酸的S原子连接成键,2-甲基二硫代丙酸分子中的SH基团与Co原子通过配位键相连;同时,Cp环的一个碳原子与碳硼烷笼体的B(3)/B(6)位相连,该B(3)/B(6)位的氢原子迁移到炔烃HC≡CCO2Me的内部炔基碳原子上形成反式烯键。3个2-甲基二硫代丙酸分子中的3个S原子分别与1中的Co原子通过共价键连接,3个SH基团与Co原子通过配位键相连,从而形成化合物3。化合物2和3分别用红外、核磁、元素分析、质谱和单晶X-射线衍射分析等方法进行了表征。
钴;碳硼烷;配体;偶联
0 Introduction
The 16e half-sandwich complexes Cp*ME2C2B10H10(M=Co,Rh,Ir;E=S,Se)and(p-cymene)MS2C2B10H10(M=Ru,Os)are stabilized as monomers by the voluminous pentamethylcyclopentadienyl(Cp*)and 1,2-dicarba-closo-dodecarborane-1,2-dichalcogenolato ligands[1-3].The combination of electron deficiency at the coordinatively unsaturated metal centre and reactivity of metal-chalcogen bonds renders these complexes promising precursors for synthesis of mixed-metal clusters[4].Besides,they could serve as interesting candidates for reactions with unsaturated substrates such as alkynes.The range of reactions turned out to be rather wide from catalytic cyclotrimerization or dimerization of the alkyne[5-6]to stepwise B(3)/(6)-substitution at the carborane cage,and numerous intermediates have been isolated[6-13].
Our previous work has indicated that the cobalt complexes CpCoS2C2B10H10and CpCoSe2C2B10H10are more reactive than their rhodium and iridium analogues[6-7].Their reactivity with methyl acetylene carboxylate leads to new complexes as the result of consecutive steps,in which the metal center and the metal-chalcogen bonds are involved as well as the BH activation at the B(3)/B(6)cite of the o-carborane cage(Scheme 1)[7].Recently,we have carried out the three-componentreaction of CpCoS2C2B10H10(1),HC≡CCO2Me and pyrrolidine-1-carbodithioic acid[14],which afforded selective B-functionalization at the carborane cage with Cp as a functional group.Herein,we report the three-component reaction of 1,HC≡CCO2Me and 2-methylpropanedithioic acid.One cobalt-promoted BH and C-H activation product and one carborane-free cobalt(Ⅱ)complex have been isolated.

Scheme 1 Some examples of complexes from the reactions of CpCoS2C2B10H10(or CpCoSe2C2B10H10)with HC≡CCO2Me[7]
1 Experimental
1.1 Reagents and instruments
All reactions and manipulations were performed under an argon atmosphere using standard Schlenk techniques.Solvents were dried by refluxing over sodium(petroleum ether,ether,and THF)or calcium hydride(CH2Cl2)under nitrogen and then distilled prior to use.CpCo(CO)I2[15]and CpCoS2C2B10H10(1)[16]were prepared according to the reported procedures. Elemental analyses were performed in an Elementar Corporation Vario ELⅢelemental analyzer.NMR data were recorded on a Bruker DRX-500 spectrometer.1H NMR and13C NMR spectra were reported with respect to CHCl3/CDCl3(δ1H=7.24,δ13C=77.0) and11B NMR spectra were reported with respect to external Et2O·BF3(δ11B=0).The IR spectra were recorded on a Bruker Tensor 27 spectrophotometer with KBr pellets in the 4 000~400 cm-1region.The mass spectra were recorded on Micromass GC-TOF for EI-MS(70 eV).
1.2 Synthesis of 2 and 3
HC≡CCO2Me(210.2 mg,2.5 mmol)and 2-methylpropanedithioic acid(120.2 mg,1.0 mmol)were added to the solution of 1(165.2 mg,0.5 mmol)in CH2Cl2(25 mL),and the mixture was stirred for 24 h at room temperature to give a deep green solution. After removing the solvent,the residue was purified on the thin layer chromatography gel and elution with petroleum ether/CH2Cl2(2∶1)gave 2 and 3(Scheme 2). Recrystallization from CH2Cl2/petrol ether afforded crystals of 2 and 3.
2:blue solid,yield 165.1 mg(62%based on 1), m.p.202℃dec.1H NMR(CDCl3):δ3.12(s,3H, Me),3.14(s,3H,Me),3.80(s,3H,OMe),4.85(s,1H, Cp-C H),5.28(s,1H,Cp-C H),5.81(s,1H,Cp-C H),5.97(s,1H,Cp-C H),6.30(d,J=16 Hz,1H,S-C H=), 7.95(d,J=16 Hz,1H,=C H-C(O)).13C NMR:δ38.06 (Me),51.98(OMe),83.30(Cp-C H),83.97(Cp-C H), 84.36(Cp-C H),92.28(Cp-C H),89.71(carborane), 117.04(carborane),120.48(S-C H),126.25(br,Cp-C), 128.20(Me2C=),143.09(=C H-C(O)),164.55(C=O), 203.05(=C S2).11BNMR(CDCl3):δ-12.7(1B),-9.3(4B), -5.3(2B),-1.7(3B).EI-MS:m/z 533.1(M+,26%).IR (KBr,ν/cm-1):1 670(C=O),1 717(S-C-S),2 582 (B-H).Elemental analysis:Calcd.for C15H25B10CoO2S4(%):C,33.82;H,4.73.Found(%):C,34.05;H,4.57.
3:green solid,yield 16.6 mg(8%based on 1), m.p.223℃dec.1H NMR(CDCl3):δ3.27(s,18H, Me).13C NMR:δ37.66(Me),134.58(Me2C=),205.41 (=C S2).EI-MS(70 eV):m/z 416.2(M+,6%).IR(KBr,ν/ cm-1):1 729(S-C-S),1 523(C=C).Elementalanalysis: Calcd.for C12H21CoS6(%):C 34.59,H 5.08.Found(%): C 34.82,H 4.87.

Scheme 2 Synthesis of 2 and 3
1.3 X-ray crystal structure determination
X-ray crystallographic data were collected on a Bruker SMART ApexⅡCCD diffractometer using graphite-monochromated Mo Kα(λ=0.071 073 nm) radiation.The intensities were corrected for Lorentz polarization effects and empirical absorption with the SADABS program[17].The structures were solved by direct methods using the SHELXS-97 program[18]and refined by full-matrix least-squares techniques on F2with the SHELXL-97 program[19].All non-hydrogen atoms were refined anisotropically.Allhydrogen atoms were generated geometrically and refined isotropically using the riding model.Crystal data,data collection parameters and the results of the analyses of 2 and 3 are listed in Table 1.Selected bond lengths and bond angles are listed in Table 2.
CCDC:1053736,2;1053737,3.
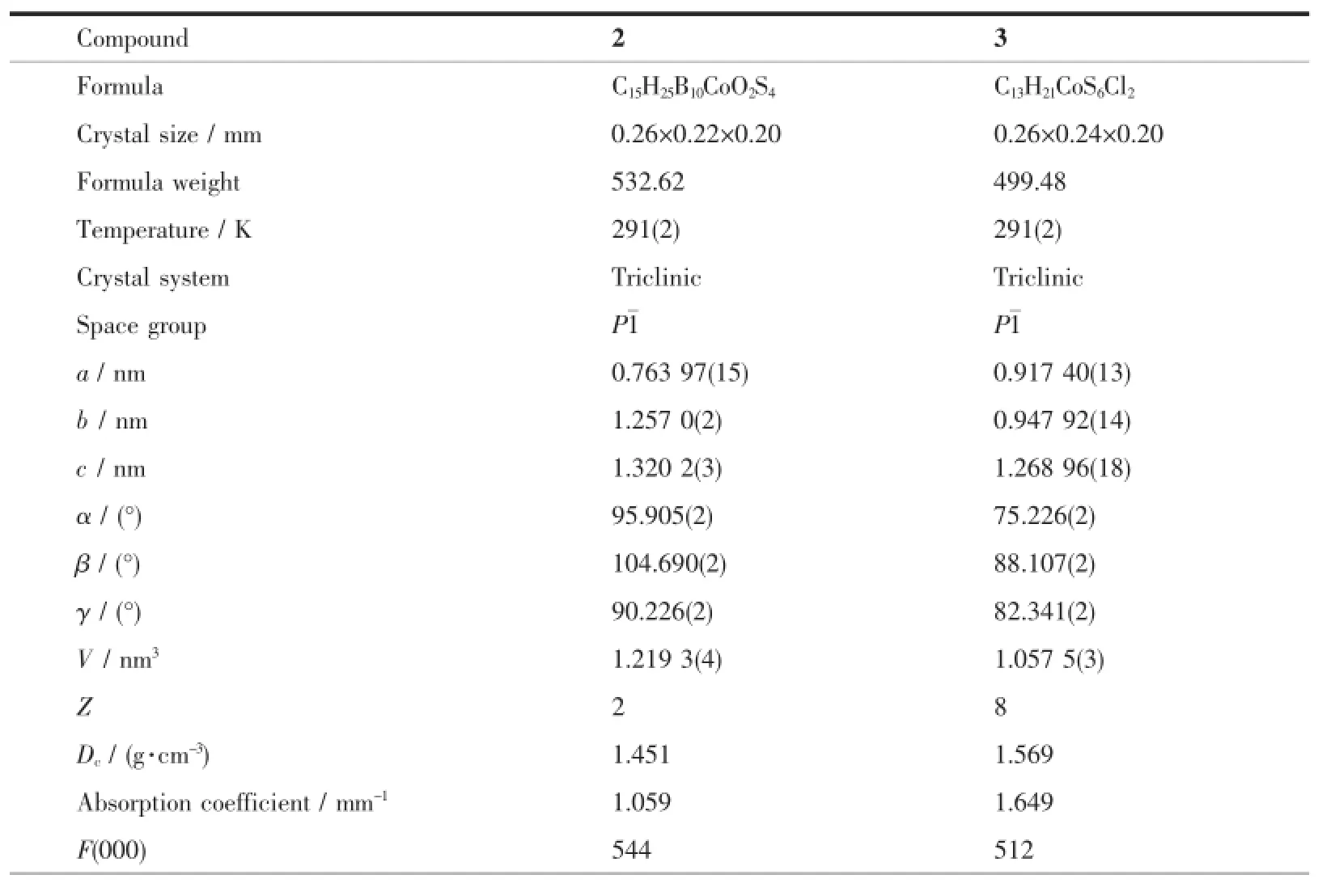
Table 1 Crystal and structure refinement data for 2 and 3

Continued Table 1

Table 2 Selected bond lengths(nm)and bond angles(°)for 2 and 3
2 Results and discussion
2.1 Structure of 2
The X-ray structure of 2(Fig.1)displays a B-C bond bridging the carborane cage and the Cp ligand. The B-C bond distance(0.156 9(6)nm)is much the same as the B-C length in trimethylborane(0.157 8 nm)[20].S(2)has disconnected from cobalt center and then connected with the S-CH=CHCO2Me unit.The newly generated five-membered ring(Co(1)S(2)C(2)B (3)C(11))is almost perpendicular to the Cp ring and fused with CpCo unit at Co(1)-C(11)bond resulting in a relative rigid Cp ring.On the other hand,the solidstate structure bears an auxiliary ligand,2-methylprop -1-ene-1,1-dithiol anion,rearranged from 2-methylpropanedithioic acid in a bidentateκ2-S,S′mode.An importantchange is thatthe C(7)-C(8)bond(0.132 0(5) nm)has been shortened,which indicates that it′s a C=C bond.Furthermore,the hydrogen atom atthe B(3)position of the carborane cage has been transferred to the terminal carbon of the alkyne to generate an olefinic substituent in a trans arrangement at the S(1) atom[14].The torsion angle of S(1)C(1)C(2)S(2)(8.7°) doesn′tchange too much as compared with 9.7°in 1[12].

Fig.1 Molecular structure of 2 with 30%thermal ellipsoids
Considering the charge compensation in the environment of cobalt center,the hydrogen atom bounded to the S(4)atom was located by constrained method.Thus,the S(4)→Co coordinative bond and two other S-Co covalent bond,together with aη5-C5H5ligand allow the metal to attain 18-electron configuration.This is consistent with the observed properties of 2.2 is a neutral compound and exhibits normal NMR spectra with no evidence of paramagnetism.However, spectroscopic data are notconsistentwith the structure in Fig.1,since there is no signature for the S-H functional group in1H NMR spectra.This implies the presence of an“extra”hydrogen atom in the vicinity of Co(1),probably bridging between the metal and one or more nearby sulfur atoms and completing an 18-electron metal configuration.The possibility of these extra hydrogens variably adopting M-H-S bridging (η2),H-MS2face-bridging(η3),or M-Hterminal(η3)modes is convinced since instead ofsulfur such bonding types for boron atoms are a well-known feature in small metallaborane and metallacarborane clusters(especially those Co,Fe and Ru clusters)[21-25].
With above comprehensible exception,spectroscopic and analytical data of 2 are consistent with its solid-state structure.In the1H NMR data,four distinguishable signals assigned to the hydrogen atoms of the substituted Cp ring could be attributed to their inequivalent chemical environment.The1H NMR spectrum also shows three singlets assigned to OMe (3.80)and Me2C(3.14,3.12)groups as well as two doublets(6.30,7.95)owing to3J(1H,1H)across the C= C bond.The large coupling constant(J=16 Hz)corresponds to an E configuration as confirmed by the solid-state structure.Characteristic resonances for CH units of the substituted Cp ring at about 83.30,83.97, 84.36 and 92.28 were found in the13C NMR spectra.
2.2 Structure of 3
The single-crystal X-ray diffraction(Fig.2)shows that3 is a neutral complex.The cobaltatom is located at the apparent intersection of D3symmetry and surrounded by six sulfuratoms in a distorted octahedral environment.Despite the apparent presence of the rotational axes,all of the atoms in the structure were found to be unique;there are no symmetry-related atoms.The average distance of three carbon-carbon bonds(C(1)-C(2),C(3)-C(4)and C(5)-C(6))is 0.131 nm,showing that they are double bonds[7].Recently,a similar compound has been synthesized in the twocomponent reaction of 1 with 2-methylpropanedithioic acid[26].However,three carbon-carbon bonds are single bonds rather than double bonds.

Fig.2 Molecular structure of 3 with 30%thermal ellipsoids
For the same reason explained for the structure of the complex 2,three hydrogen were constrainedly located at S(2),S(4)and S(5),respectively,and there were no signals in1H NMR spectra.However,the spectroscopic and analytical data support its principal part in the solid-state structure.The singlet at 3.27 was assigned to the alkyl group of the Me2C unit as expected for a symmetricalstructure.
The work described here sheds light on a relatively unexplored facet of o-carborane chemistry. Further experimental and theoretical work is underway.
[1]Herberhold M,Jin G X,Yan H,et al.Eur.J.Inorg.Chem.,1999,5:873-875
[2]Herberhold M,Jin G X,Yan H,et al.J.Organomet.Chem., 1999,587:252-257
[3]Bae J Y,Park Y L,Ko J,et al.Inorg.Chim.Acta,1999,289: 141-148
[4]Liu S,Han Y F,Jin G X.Chem.Soc.Rev.,2007,36:1543-1560
[5]Herberhold M,Yan H,Milius W,et al.Organometallics, 2000,19:4289-4294
[6]Xu B H,Wu D H,Li Y Z,et al.Organometallics,2007,26: 4344-4349
[7]Xu B H,Tao J C,Li Y Z,et al.Organometallics,2008,27: 334-340
[8]Herberhold M,Yan H,Milius W,et al.Chem.Eur.J.,2000, 6:3026-3032
[9]Herberhold M,Yan H,Milius W,et al.J.Chem.Soc.,Dalton Trans.,2001:1782-1789
[10]Herberhold M,Yan H,Milius W,et al.Angew.Chem.Int. Ed.,1999,38:3689-3691
[11]Herberhold M,Yan H,Milius W,etal.Z.Anorg.Allg.Chem., 2000,626:1627-1633
[12]Kim D H,Ko J,Park K,et al.Organometallics,1999,18: 2738-2740
[13]Won J H,Kim D H,Kim B Y,et al.Organometallics,2002, 21:1443-1453
[14]Zhang R,Zhu L,Liu G F,et al.J.Am.Chem.Soc.,2012, 134:10341-10344
[15]Frith S A,Spencer J L.Inorg.Synth.,1990,28:273-280
[16]Hou X F,Wang X,Wang J Q,et al.J.Organomet.Chem., 2004,689:2228-2235
[17]Sheldrick G M.SADABS,An EmpiricalAbsorption Correction Program,Bruker Analytical X-ray Systems,Madison,WI, 1996.
[18]Sheldrick G M.SHELXS-97,Program for Crystal Structure Solution,University of Göttingen,Göttingen,Germany, 1997.
[19]Sheldrick G M.SHELXL-97,Program for Crystal Structure Refinement,University of Göttingen,Göttingen,Germany, 1997.
[20]Boese R,Blaser D,Niederprum N,et al.Angew.Chem.Int. Ed.,1992,31:314-316
[21]Yao H J,Grimes R N.Organometallics,2003,22:4539-4546
[22]Russell J M,Sabat H,Grimes R N.Organometallics,2002, 21:5613-5621
[23]Yan H,Beatty A M,Fehlner T P.Angew.Chem.Int.Ed., 2001,40:4498-4501
[24]Yan H,Beatty A M,Fehlner T P.Angew.Chem.Int.Ed., 2002,41:2578-2581
[25]Yan H,Beatty A M,Fehlner T P.J.Am.Chem.Soc.,2002, 124:10280-10281
[26]Zhang R,Zhu L,Lu Z Z,et al.Dalton Trans.,2012,41: 12054-12063
Cobalt(Ⅱ)-Mediated Intramolecular Coupling of B(3)/B(6)in CpCoS2C2B10H10with Cp Ligand
YE Hong-De*,1,2XU Bao-Hua1HU Jiu-Rong2YAN Hong*,1
(1State Key Laboratory of Coordination Chemistry,School of Chemistry and Chemical Engineering,Nanjing University,Nanjing 210093,China) (2School of Chemistry and Chemical Engineering,Shangrao Normal University,Shangrao,Jiangxi 334001,China)
Half-sandwich 16e complex CpCoS2C2B10H10(Cp:cyclopentadienyl)(1)reacted with HC≡CCO2Me in the presence of 2-methylpropanedithioic acid to generate compounds{(C5H4CoS2C2B9H9)(CH=CHCO2Me)(Me2C= CS2H)}(2)and(Me2C=CS2H)3Co(3).In 2,one S-Co bond of 1 was broken,and the S atom linked with the terminal acetylenyl carbon atom of the HC≡CCO2Me.Meanwhile,the Co atom in 1 linked to the S atom in 2-methylpropanedithioic acid with covalent bond while linked to the SH unit with coordinative bond.One carbon atom of Cp ring linked to the B(3)/B(6)cite of the carborane cage.The H atom of the B(3)/B(6)cite migrated to the internal acetylenyl carbon atom to form a trans-ethylenic bond.The Co atom of 1 linked to three S atoms of three 2-methylpropanedithioic acid molecules with covalent bond,while linked to the other three SH units with coordinative bond.As a result,product 3 formed.Complexes 2 and 3 have been characterized by IR,NMR, elemental analysis,mass spectrum and single-crystal X-ray diffraction analysis.CCDC:1053736,2;1053737,3.
cobalt;carborane;ligand;couple
O614.81+2
A
1001-4861(2015)07-1447-06
10.11862/CJIC.2015.200
2015-03-29。收修改稿日期:2015-05-08。
国家自然科学基金(No.21361022,21261020)、第54批中国博士后科学基金面上基金(No.2013M541640)、南京大学配位化学国家重点实验室第25批开放课题资助项目。
*通讯联系人。E-mail:yehongde@163.com,hyan1965@nju.edu.cn
猜你喜欢
中学生数理化(高中版.高考理化)(2024年2期)2024-03-20 01:30:20
家庭影院技术(2021年10期)2021-11-20 06:08:50
家庭影院技术(2020年8期)2020-09-11 06:44:44
家庭影院技术(2020年3期)2020-05-21 07:28:02
中小学班主任(2019年12期)2019-09-10 07:22:44
天然产物研究与开发(2018年5期)2018-06-13 03:23:46
中学生数理化·高二版(2017年2期)2017-04-19 16:29:54
科技创新导报(2016年1期)2016-05-30 06:38:19
中成药(2016年8期)2016-05-17 06:08:36
化工进展(2015年3期)2015-11-11 09:09:40
