
HPLC法测定百仙妇炎清凝胶剂中苦参碱的含量
2015-11-29 11:44:47韩忠耀鲁利民陆锦锐
中国中医药现代远程教育 2015年6期
韩忠耀鲁利民陆锦锐*
(1黔南民族医学高等专科学校药学系,都匀558000;2中国中医科学院西苑医院药剂科,北京100091)
HPLC法测定百仙妇炎清凝胶剂中苦参碱的含量
韩忠耀1鲁利民2陆锦锐1*
(1黔南民族医学高等专科学校药学系,都匀558000;2中国中医科学院西苑医院药剂科,北京100091)
目的建立百仙妇炎清凝胶剂中苦参碱的含量测定方法。方法采用HPLC法测定百仙妇炎清凝胶剂中苦参碱的含量,选用Promosil C18色谱柱(250 mm×4.6 mm,5 μm),检测波长210 nm,流动相:0.02 mol/L磷酸二氢钾试液-0.2%三乙胺:乙腈(85:15),流速1.0 mL/min,柱温35℃。结果苦参碱在0.13~2.34 μg范围内线性关系良好(r=0.9999);平均回收率为98.35%,RSD=1.52%。结论所建立的方法准确可靠,适合于测定百仙妇炎清凝胶剂中苦参碱的含量。同时,本方法操作简便,灵敏度高,干扰少,重现性较好,可以作为百仙妇炎清凝胶剂的质量控制方法。
百仙妇炎清凝胶剂;苦参碱;高效液相色谱法;质量控制
百仙妇炎清凝胶剂是由百仙妇炎清栓改剂型而来的中药八类新药。百仙妇炎清凝胶剂主要由苦参、百部、蛇床子、仙鹤草、紫珠叶、白矾、冰片、樟脑、硼酸等药材制成的中药复方制剂。全方共奏清热解毒、杀虫止痒、去瘀等功效,主要用于霉菌性、细菌性、滴虫性阴道炎和宫颈糜烂。
改成凝胶剂后,与栓剂相比,其优点是:首先,水溶性的凝胶比栓剂使用时洁净度更高,不污染衣物;其次,局部药物浓度高,与病灶接触面积大,能够全面覆盖阴道粘膜,不留死角;然后,阴道内给药,直接作用于患处;同时,该剂型给药方便,阴道内滞留性好,药物作用时间长,无刺激,无异物感。通过处方制剂等研究,产品稳定,质量可控、安全、使用与携带方便,证明所选的剂型合理可行。
目前百仙妇炎清凝胶剂研究报道中[1-6],尚无针对百仙妇炎清凝胶剂方中君药苦参的主要活性成分苦参碱的含量控制的研究。本研究结合2010版(《中国药典》一部)[7]与相关文献,旨在探讨百仙妇炎清凝胶剂中苦参碱的含量测定方法,为百仙妇炎清凝胶剂制定质量标准提供基础与依据。
1 仪器与试药
1.1 仪器Agilent1100型高效液相色谱仪(美国安捷伦科技公司);博纳艾杰尔Promosil C18色谱柱(250mm× 4.6mm,5μm);Sartorius BP211D型分析天平(德国Sartorius公司)。
1.2 试药苦参碱对照品(中国药品生物制品检定所,批号:110805-200507);百仙妇炎清凝胶(批号:20131201、20131202、20131203,实验室自制);色谱纯乙腈(天津康科德科技有限公司);磷酸为分析纯,水为超纯水,其他试剂均为分析纯。
2 实验方法与结果
2.1 色谱条件流动相:0.02 mol/l磷酸二氢钾试液-0.2%三乙胺:乙腈(85∶15);检测波长:210 nm;流速1.0 mL/min;柱温35℃;进样量:10 μL。理论板数按苦参碱峰(C15H24N2O)计算应不低于2000。
2.2 对照品溶液的制备取苦参碱对照品,加甲醇溶解,制成每1 mL含苦参碱0.26 mg的溶液,作为对照品贮备溶液。
2.3 供试品溶液的制备取样品适量,精密称定百仙凝胶剂0.5 g,置于25 mL量瓶中,加甲醇24 mL,超声提取30分钟,放冷,加甲醇稀释至刻度,摇匀,0.45 μm微孔滤膜滤过,弃去初滤液,取续滤液,作为供试品溶液。
2.4 阴性样品溶液的制备按处方比例及工艺制备缺苦参药材的阴性对照品溶液。
2.5 专属性试验分别吸取对照品溶液、阴性样品溶液及百仙妇炎清凝胶供试品溶液,进样10 μL,按上述色谱条件测定,进行HPLC分析,结果阴性对照品溶液在相应的保留时间处无色谱峰,表明其它组分对测定无干扰,见图1,1为苦参碱峰。
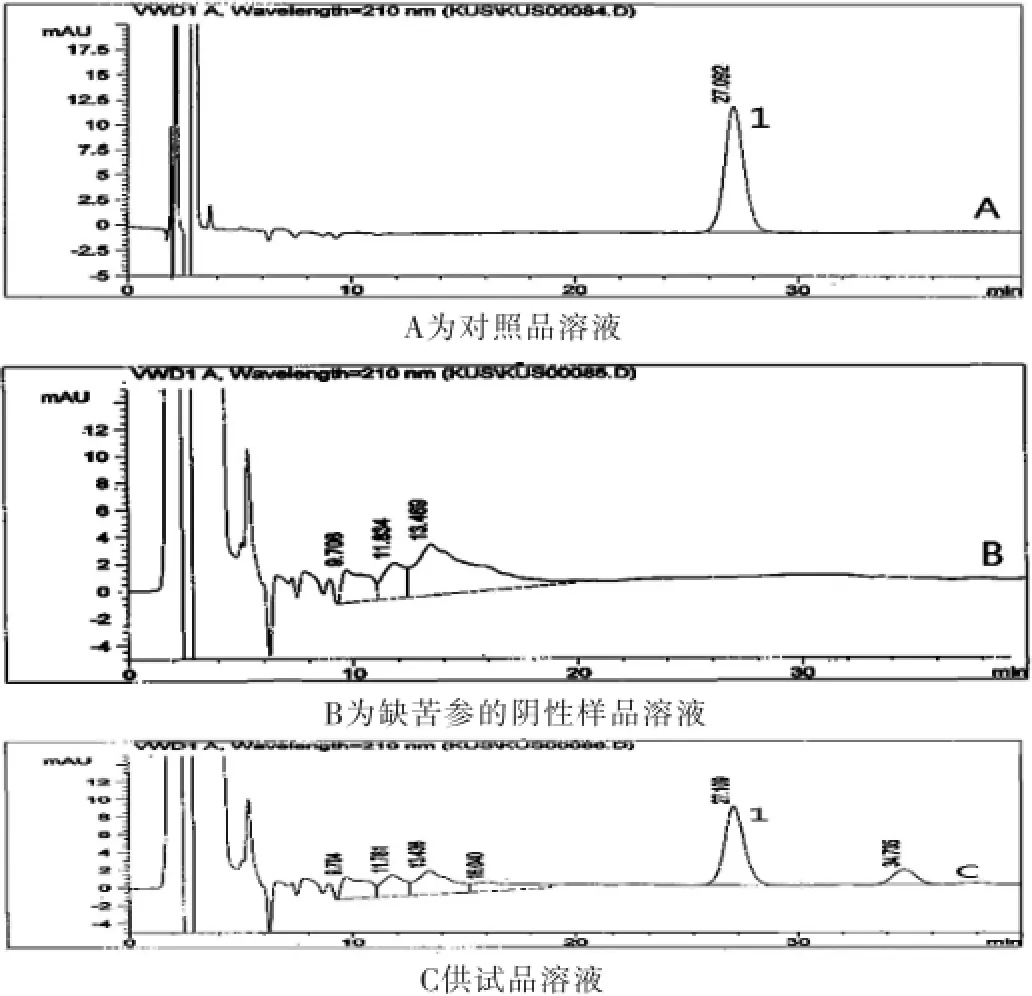
图1 百仙妇炎清凝胶HPLC
2.6 线性关系的考察精密吸取苦参碱对照品溶液(0.26 mg·mL-1)0.5,1,3,5,7,和9 mL,置于6个10 mL量瓶中,分别用甲醇定容至刻度;在上述色谱条件下,分别进样10 μL,以进样量为横坐标,峰面积为纵坐标,绘制标准曲线,计算,得回归方程Y=405.72X+17.145,r=0.9999。结果表明苦参碱进样量在0.13~2.34 μg·mL-1范围内呈良好的线性关系。
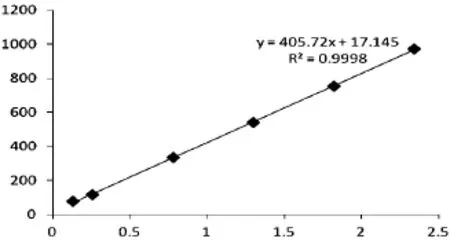
图2 苦参碱线性考察
2.7 精密度试验取对照品溶液,按上述色谱条件测定,连续进样6次,测定峰面积RSD为0.75%。
2.8 稳定性实验取供试品溶液分别在0,4,8,12,24 h进样,测定峰面积,计算得RSD为1.29%。结果表明供试品溶液在24 h内稳定。
2.9 重现性试验取同一批制剂,按供试品溶液制备方法制备6份溶液,在上述色谱条件下测定,计算苦参碱含量,RSD为0.94%。
2.10 回收率试验采用加样回收法,精密称取已知含量的本品半量约0.25 g,置25 mL量瓶中,分别精密吸取对照品溶液(浓度0.0999 mg/ml,3.5,6,8.5 mL,各3份,加入含样品0.25 g的量瓶中,(加入对照品的量相当于全量的80%、100%、120%),按含量测定项下,分别测定苦参碱的含量,进样量10 μL,记录色谱峰,计算回收率。结果见下表。

表1 加样回收率考察表
2.11 含量测定百仙妇炎清凝胶剂三批样品(批号:20131201、20131202、20131203,实验室自制)测定按上述色谱条件对3批样品进行分析,以外标法计算苦参碱含量,含量测定结果如下:

表2 百仙妇炎清凝胶含量检测结果
3 讨论
赵玉绒[2]等采用NH2色谱柱测定百仙妇炎清阴道泡腾片中苦参碱的含量,由于NH2色谱柱价格昂贵,较特殊,所以将其改成价格低廉、常用的C18色谱柱,为检测提供便利条件,更符合实际工作需要。汪暐东[3]以乙腈-0.025 mol×L-1硫酸铵-十二烷基硫酸钠(30:70: 0.6)(用硫酸(1→5)溶液调节pH至3.0)为流动相,梅璇[4]等以磷酸缓冲溶液(0.2%三乙胺水溶液1000 mL,用磷酸调pH 3.5)-乙腈(96∶4)为流动相,均需用酸调节溶液的pH,操作较复杂,本文为流动相:0.02 mol/L磷酸二氢钾试液-0.2%三乙胺:乙腈(85∶15),实验操作简单,方便。
本品在进行波长选择时,称取苦参碱对照品适量,用甲醇溶解后做紫外扫描,测得最大吸收波长为201.9 nm,因此,波长有末端吸收,有干扰,参照结果最终选定210 nm作为百仙妇炎清凝胶剂检测波长。
3.1 提取溶剂考察取本品(批号:20131201)4份,置于25 mL量瓶中,分别加入甲醇、无水乙醇、2%氨性氯仿、流动相约24 mL,超声处理30分钟后,取出,放冷,加提取溶剂稀释至刻度,摇匀,用0.45 μm膜滤过,弃去初滤液,取续滤液为供试品溶液,分别进样10 μL,记录色谱图,各提取溶剂下苦参碱含量测定结果见表3。

表3 提取溶剂考察结果表
由以上结果可见,苦参碱含量大小顺序依次为:甲醇提取>无水乙醇提取>2%氨性氯仿提取。同时,流动相提取的样品,由于凝胶剂中辅料的溶解而难以滤过,所以最终确定甲醇为本品的提取溶剂。
3.2 提取方法考察
3.2.1 超声提取法取本品(批号:20131201),置25 mL量瓶中,加甲醇约24 mL,超声处理30分钟,取出,放冷,加甲醇稀释至刻度,摇匀,用0.45 μm膜滤过,弃去初滤液,取续滤液为供试品溶液,进样10 μL。
3.2.2 回流提取法取本品(批号:20131201),置50 mL三角瓶中,精密加入甲醇25 mL,称重,水浴加热回流提取30 min,放冷,称重,补足甲醇,摇匀,用0.45 μm膜滤过,弃去初滤液,取续滤液为供试品溶液,进样10 μL。结果见表4。

表4 提取方法考察结果表
由以上结果可见,超声提取的苦参碱含量高于回流提取,且方法简便,所以确定以超声提取为本品的提取方法。
3.2.3 提取时间考察取本品(批号:20131201)3份,置25 mL量瓶中,加甲醇约24 mL,分别超声处理10、 30、60 min,使溶解,取出,放冷,加甲醇稀释至刻度,摇匀,用0.45 μm滤膜过滤,弃去初滤液,取续滤液为供试品溶液,进样10 μL,结果见表5。

表5 提取时间考察表
由以上结果可见,超声提取30 min已提取完全,检测到的苦参碱的含量几乎无变化,表明此时苦参碱已经提取彻底,所以确定以超声30 min为本品的提取方法。
将百仙妇炎清栓改制成百仙妇炎清凝胶,可减少刺激性,更便于临床应用。通过HPLC法测定百仙妇炎清凝胶中的君药苦参的主要活性成分苦参碱进行含量测定,可为百仙妇炎清凝胶质量控制及制定质量标准提供基础与借鉴。
[1]程雪翔.HPLC测定百仙妇炎清凝胶中蛇床子素的含量[J].中国中药杂志,2007,32(12):1227-1229.
[2]赵玉绒,郭永逹.百仙妇炎清阴道泡腾片的质量标准研究[J].西北药学杂志,2009,24(6):460-462.
[3]汪暐东.HPLC测定百仙妇炎清栓中的苦参碱含量[J].安徽医药,2008,12(12):1154-1155.
[4]梅璇,刘致珍.反相高效液相色谱法测定百仙妇炎清栓中的苦参碱含量[J].贵州医药,2006,30(10):937-938.
[5]梁艺英,朱炳辉,方继辉,等.苦参碱葡萄糖注射液中苦参碱的含量测定[J].中国现代应用药学杂志,2004,21(5):399-401.
[6]刘庆丰,李中东,施孝全,等.高效液相色谱法测定除湿注射剂中的三种成分[J].中国现代应用药学杂志,2007,24(5):412-413.
[7]国家药典委员会.中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2010:188.
Content Determination of Matrine in Baixian Fuyanqing Ningjiao by HPLC
HAN Zhongyao1,LU Limin2,LU Jinrui1*
(1 Department of Pharmacy,Qiannan Medical College for Nationalities,Guizhou Province,Duyun558000,China;2 Pharmacy Department,Xiyuan Hospltal CACMS,Beijing100091,China)
Objective Establish the method for the content determination of matrine in Baixian Fuyanqing Ningjiao.Methods The matrine content of Baixian Fuyanqing Ningjiao was determined by HPLC with Promosil C18 column(250 mm×4.6 mm,5 μm).The mobile phase was 0.02 mol/L Potassium dihydrogen phosphate-0.2% triethylamine:acetonitrile(85∶15),and the detection wavelength was 210 nm,the flow rate was 1 mL/min,the column temperature was 35℃.Results The calibration curve was linear over the range of 0.13~2.34 μg(r=0.9999)for matrine.The average recovery rate was 98.35%,RSD=1.52%.Conclusion This method has good characteristics and is suitable for the content determination of matrine in Baixian Fuyanqing Ningjiao.Meanwhile,this method is simple,sensitive and reproducible.It can be used for the quality control of the Baixian Fuyanqing Ningjiao.
Baixian Fuyanqing Ningjiao;matrine;HPLC;quality control
10.3969/j.issn.1672-2779.2015.06.071
1672-2779(2015)-06-0140-03
杨杰 本文校对:杨杰
2015-01-30)
*通讯作者:lujinrui89@126.com
猜你喜欢
环境卫生工程(2021年3期)2021-07-21 05:34:36
健康之家(2021年19期)2021-05-23 11:17:03
甘肃医药(2020年10期)2021-01-07 03:35:58
云南医药(2020年5期)2020-10-27 01:37:58
环境卫生工程(2020年3期)2020-07-27 01:19:18
供水技术(2020年6期)2020-03-17 08:18:22
中成药(2018年12期)2018-12-29 12:25:54
天然产物研究与开发(2018年5期)2018-06-13 03:23:34
中国民族医药杂志(2016年5期)2016-05-09 07:43:50
环境科技(2015年2期)2015-11-08 12:11:24
