
芬戈莫德研发历程概述
2015-11-18 04:25:15钮俊兴胡立宏
药学研究 2015年12期
钮俊兴,胡立宏
(中国科学院上海药物研究所,中药现代化中心,上海 201203)
芬戈莫德研发历程概述
钮俊兴,胡立宏
(中国科学院上海药物研究所,中药现代化中心,上海201203)
芬戈莫德是以真菌的次级代谢产物ISPI为苗头化合物所研发的免疫调节剂,于2010年被FDA批准上市,用以治疗免疫性疾病多发性硬化症。与传统的免疫抑制剂不同,它不影响淋巴细胞的活化和增殖,主要作用于细胞表面的1-磷酸鞘氨醇(S1P)受体来发挥免疫抑制和免疫调控作用。本文通过梳理芬戈莫德的研发流程,阐述结构优化和改造步骤,为药物化学研究提供经验。
芬戈莫德;ISP-I;多发性硬化症免疫调节剂
专家简介
胡立宏,男,中国科学院上海药物研究所研究员、研究组长、博士生导师,研究工作主要涉及“基于中草药资源的药物发现”研究领域。现任世界中医药学会联合会中药分析专业委员会理事、中国植物学会民族植物学分会理事、上海市药学会天然药化专业委员会委员、上海市口腔医学重点实验室学术委员会委员,以通讯作者身份在Journal of Neuroscience,Chemistry&Biology,Journal of Medicinal Chemistry,Diabetes,Green Chemistry,Organic Letters等国际主流刊物上发表研究论文190余篇,获得发明专利授权10余项。2009年以“天然药物化学”研究方向获得“国家杰出青年基金”资助,并入选“中国科学院百人计划”,2012年获得了“上海市优秀学科带头人”称号。
1 背景
多发性硬化症(multiple sclerosis,MS)是以中枢神经系统蛋白质炎性脱髓鞘病变为主要特点的自身免疫病。目前全球约有250万人受MS困扰,多发于20~40岁青年人群,女性患者为男性两倍[1]。发病原因未知,一般认为与遗传、环境、自身免疫等因素有关。MS最常累及的部位为脑室,会造成不可逆的神经损伤,并因此产生广泛的症状,诸如肢体无力、精神异常、共济失调、视力衰退等[2]。
目前,临床上用于治疗MS的药物有免疫调节剂、免疫抑制剂和抗炎药3类[3],但这些药物对于MS的治疗疗效不显著且副作用大。因此,人们迫切需要一种疗效好,病人依从性好,副作用小的药物。而芬戈莫德(Fingolimod)的出现,为广大患者带来了福音。
芬戈莫德是来源于天然产物的、首个口服给药的用于治疗MS的新型免疫抑制剂。它通过调节神经细胞的1-磷酸鞘氨醇(S1P)受体,使淋巴细胞回流至淋巴结从而起到降低MS复发频率、延缓病程的作用[4,5]。
2 芬戈莫德的研发历程
2.1苗头化合物的发现20世纪中叶后,随着青霉素类抗生素的兴起,从细菌、真菌的代谢产物中寻找活性物质成为潮流,日本科学家藤田也不例外,他的研究对象是真菌辛克莱虫草(Cordyceps sinclairii)和其近亲冬虫夏草(Cordyceps sinensis),并在20世纪80年代末分离得到化合物ISP-I[6,7]。
ISP-I有较强的免疫抑制活性,与已有抗MS药物环孢素A(CsA)相比,体外活性是后者的5~10倍,体内活性是其10~100倍[8]。但是,该化合物的毒性比CsA大100倍,且溶解度低,成药性差;同时手性原子较多,结构相对复杂,想要成为药物用于临床还需进一步的结构优化。
因此,藤田小组决定以ISP-I作为苗头化合物(Hit),从结构简化角度入手,通过进一步的结构优化获得成药性更佳的候选化合物。
为了定量的衡量和评价所得化合物的免疫抑制活性,藤田小组设计了两种生物学模型来评价化合物的免疫抑制作用:分别是体外的T细胞活性抑制实验和体内的小鼠间皮移植实验[9]。体外试验通过测定T细胞的半数抑制浓度(IC50)来表征化合物免疫抑制活性;体内实验通过测定小鼠皮移植后平均存活时间(MST),间接评价化合物抑制活性和毒性。
2.2先导化合物的确定藤田小组在得到真菌次生代谢产物—化合物1(ISP-I)的同时,同时还得到其同系物2(Mycestericin D)、3(Mycestericin E)、4(Mycestericin F)和5(Mycestericin G)[10],其结构如表1所示。
分析表1的数据,可以得出初步的结构与活性的关系:化合物2与1比较,其少了4-OH,活性略微下降,说明4-OH为活性非必需基团,可以除去;化合物2与3活性相差不大,说明3-OH的构型与活性无关;6、7位双键还原后的化合物4和5活性显著下降,说明6,7位双键是活性必须的。
2.2.114位羰基的必要性为研究14位羰基的必要性,藤田小组设计、合成了化合物6、7,如表1所示。
比较化合物1、6、7的活性数据发现,将羰基还原为羟基活性略有下降;将羰基转变为亚甲基活性略有提升,说明羰基并非活性必须基团,将其还原为亚甲基可简化结构,并提高活性。

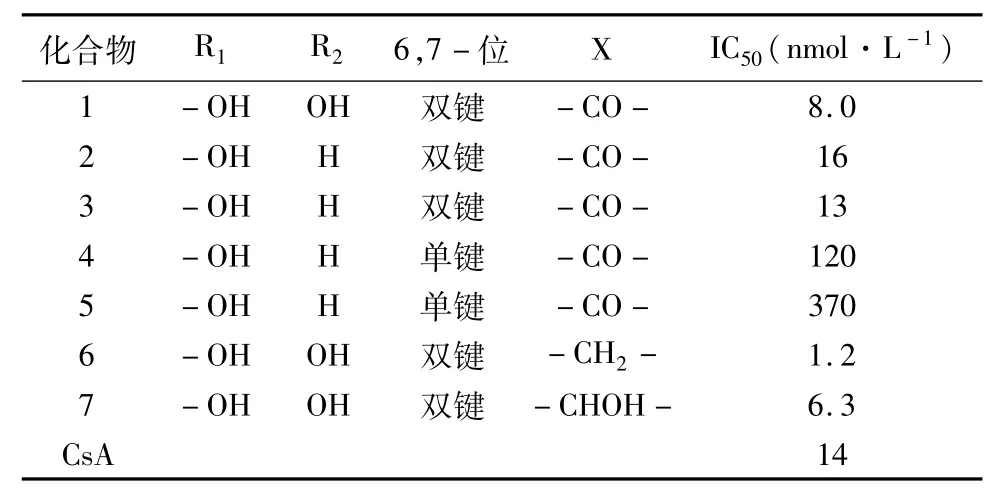
表1 化合物1~7对T细胞抑制活性数据表
2.2.22位手性碳构型与活性关系再次分析原型化合物1的结构,其2位是一个手性碳,合成难度较大,不利于进一步修饰改造;同时1又包含酸、碱两性结构和亲水、亲脂的两性结构,水溶性低。于是藤田小组又尝试探索2位手性碳构型和羧基对于活性的影响,设计并合成了一系列化合物8~11[11],见表2。

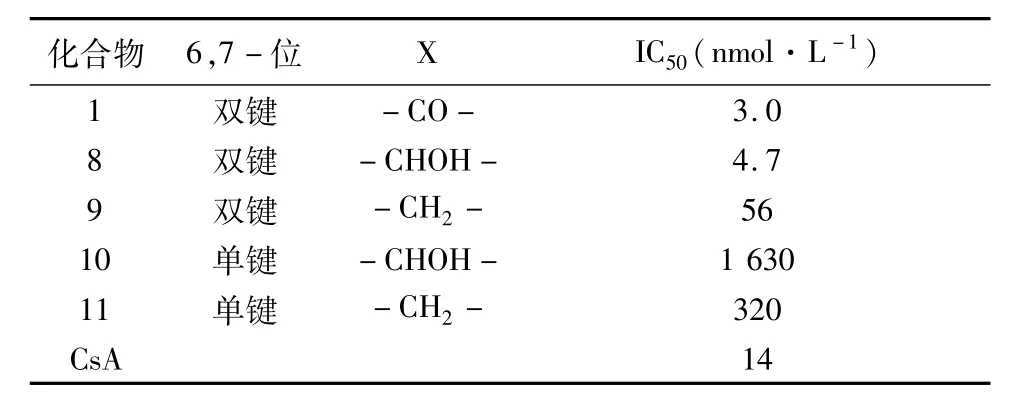
表2 化合物8~11对T细胞抑制活性数据表
比较化合物1和8发现,2位手性碳构型与活性无关,1-COOH可还原为1-CH2OH;化合物10、11的活性丧失进一步验证了对脂肪链进行构象限制对生物活性是至关重要的。
2.2.3脂肪链链长与活性关系接下来,藤田小组考察脂肪链长度与活性的关系,为此,他们设计了一系列化合物12a~12j[12],如表3所示。
分析表3的数据,发现当脂肪链碳原子数目为15(12f)或14(12e)时,化合物活性均较好;而碳原子数目大于15或小于14时,化合物的活性均显著降低;进而以口服给药方式继续比较12e和12f的体内活性(见表7),结果显示,当给药剂量为30 mg·kg-1时,CsA的MST为20.5d,12e为52.2d,而12f则表现出毒性。化合物12e口服给药下的生存期明显长于化合物12f和阳性对照药CsA,因此,可以确定脂肪链长度为14的碳原子时,其活性较好,毒性较小。

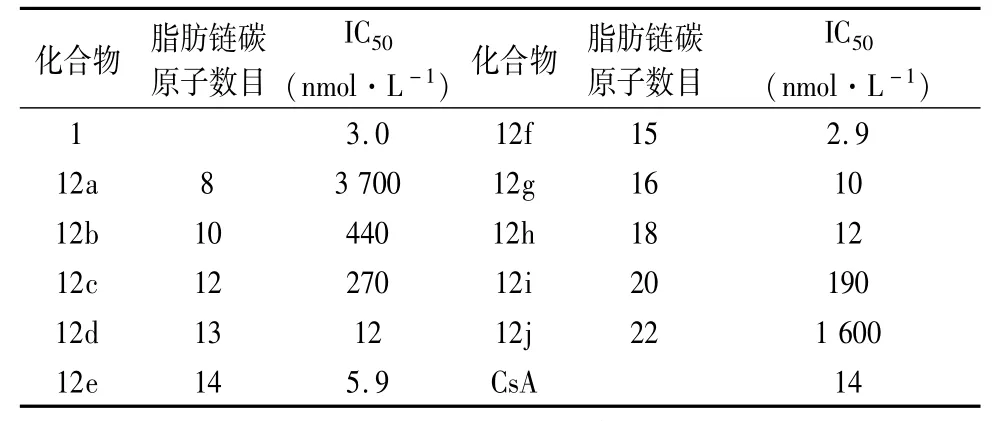
表3 化合物12a~12j对T细胞抑制活性数据表
根据以上结果,藤田小组决定以12e为先导化合物,进行进一步的结构优化。化合物12e的结构由右侧亲脂部分和左侧亲水部分组成,因此接下来将分别对这2个部分进行修饰改造。
2.3亲脂部分改造
2.3.1引入芳香环由上文已知脂肪链上6、7位双键对于化合物活性是必不可少的,分析原因可能是脂肪链柔性过大,而双键可起到限制构象以及π-π堆积的作用。于是藤田小组尝试引入苯环,在保持脂肪链碳原子总数为14的同时,获得了苯环处于不同位置的系列化合物14~21,见表4。


表4 化合物13~21对T细胞抑制活性数据表
分析表4的数据,发现化合物16活性较好,证明猜想正确:即引入苯环可以限制构象,提高化合物生物活性;同时也得知芳香环与亲水部分之间间隔2个碳原子的距离为佳。
2.3.2苯环右侧脂肪链链长与活性关系由上文已知,引入苯环可提高活性,且环与亲水部分需间隔2个碳原子。随后,藤田小组又合成了一系列化合物22~28,用以确定苯环右侧脂肪链长度与活性的关系,如表5所示。


表5 化合物22~28对T细胞抑制活性表
分析表5的数据,发现在保证苯环与亲水部分间隔两个碳原子的前提下,化合物活性对苯环上的脂肪链取代基长度不明感,因为化合物22~28大多具有良好的生物活性。其中活性最好的化合物23因体内活性弱于化合物16(见表7),故藤田小组决定保持苯环上连接正辛基,即以化合物16继续进行改造。
2.3.3苯环与脂肪链连接部分改造接着,藤田小组又合成了化合物29~34,尝试对苯环和脂肪链的连接部分进行改造,化合物结构与活性如表6所示。

表6 化合物29~34对T细胞抑制活性表
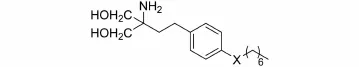
分析表6的数据,发现化合物29有与16相当的活性,但其在体内中,3 mg·kg-1情况下显示出毒性(见表7);引入氧原子的32也具有与16相当的活性,但MST比16短;引入硫原子所得的33活性也很好,遗憾的是毒性也随之增大;引入含氮基团所得的31与34活性较差。
以上结论说明苄位的改造不利于化合物活性的提高和毒性的降低。
2.3.4苯环上脂肪链取代基位置确定随后藤田小组合成了化合物35、36,用来确定苯环上脂肪链取代基位置对于化合物活性的影响,如表8所示。

表7 相关化合物小鼠皮移植存活时间表
分析表8的数据发现脂肪链取代基位于苯环对位时活性最高,而位于邻位、间位时活性丧失。
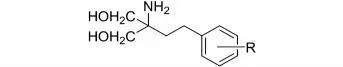

表8 取代基位置与T细胞抑制活表
综合以上结果,化合物12e亲脂部分的结构优化得到药物候选物16。
2.4亲水部分改造接下来藤田小组对先导化合物12e的亲水部分也进行了结构优化。考虑到“2.3.3”中苄位引入氧原子有可能提高化合物生物活性,于是,设计了相关化合物37~47,分别考察苄位原子和亲水部分的取代基对于化合物活性的影响,结构与活性如表9所示。
分析表9的数据可知,参照ISP-I结构将羟甲基氧化为羧基、或直接去除都会使化合物活性大大下降(化合物37、38);将羟甲基链长伸长也会使活性小幅度下降(化合物45~47),说明羟甲基不是一个合适的改造位点;而在苄位引入氧原子的情况下,化合物39~45的体外活性相对于16均有一定程度的下降,说明苄位引入杂原子不利于生物活性的保持。

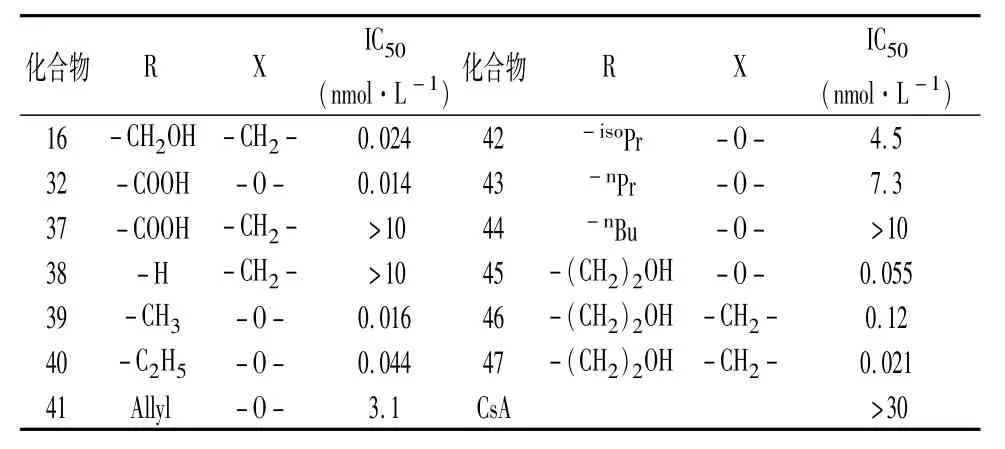
表9 化合物37~47对T细胞的抑制活性
综合以上结果,藤田等选择化合物16(FTY720)作为候选化合物进行临床研究,在顺利通过Ⅰ期、Ⅱ期、Ⅲ期临床后,于2010年被FDA批准上市,用于治疗MS,这也是首个口服给药的用于治疗MS的新型免疫抑制剂。
3 芬戈莫德治疗MS机制研究
Brinkmann等[1]对芬戈莫德的抗MS机制进行了细致的研究,得到了有趣的结果。
鞘氨醇(sphingosine)被鞘氨醇激酶2磷酸化形成S1P(见图1)是产生T细胞与B细胞级联反应的起始步骤。而S1P是5个不同的G蛋白偶联受体(GPCRs)的激活剂,这些G蛋白偶联受体的激活是淋巴结淋巴细胞在体内释放的必要过程[13,14]。
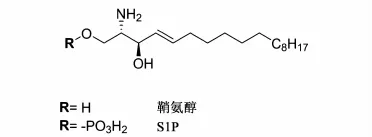
图1 鞘氨醇和S1P结构图
淋巴细胞的过度释放,会导致诸如MS、内部炎症、细胞凋亡等多种病理过程。目前MS的治疗药物有CsA和他克莫司(FK506),其机理是通过抑制丝氨酸棕榈酰转移酶从而抑制鞘氨醇的生物合成,使相关的S1P受体1~5不能激活,最终抑制T细胞和B细胞的释放,使身体不能产生任何免疫反应,起到缓解MS的作用。
而抗MS新药芬戈莫德(Fingolimod)具有独特和新颖的作用机制:芬戈莫德一旦吸收入血,将迅速被鞘氨醇激酶2磷酸化成磷酸化芬戈莫德(见图2),后者与内源性配体S1P具有结构相似性,因而能够竞争性的结合到 S1P受体上,起到阻断 GPCRs信号传导的作用[4,15]。母体芬戈莫德没有任何受体结合能力,体内药代动力学研究也已证实,口服芬戈莫德后,磷酸化芬戈莫德的血药浓度是其母体的4倍[16,17],这也是其得以发挥药理活性的基础。
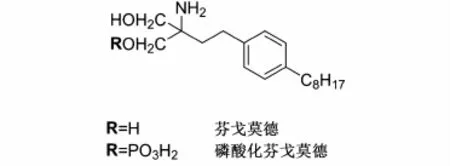
图2 芬戈莫德和磷酸化芬戈莫德结构图
4 总结
藤田在发现天然产物ISP-I的免疫抑制活性后,通过分析确定了以化合物12e作为先导化合物,进行了进一步的结构修饰与改造,获得活性化合物16(FYP720,芬戈莫德),并最终于2010年被FDA批准上市[18],它的优化过程对我们启示颇多。
4.1简化结构、去除手性原子。最初发现的天然产物ISP-I具有3个手性中心,结构较为复杂,合成困难,不利于进一步研究。藤田组首先就探索了手性碳原子的必要性,在确定了其不影响活性后,改用去手性衍生物进行后续探索。利用药物改造的基本思想,在进一步简化了结构的同时,提高了化合物活性,降低了合成难度和成本,使得后续的大规模生产成为可能。
4.2增加溶解度,降低毒性。前体化合物ISP-I的水溶解性很低且毒性大,因此藤田小组在保证活性的基础上,通过生物电子等排、保持药效团等手段改善了化合物的水溶性,同时使毒性降低到可接受范围内,推动了其走向临床的过程。
4.3合理的筛选模型。芬戈莫德是在受体未知的情况下,基于药物化学理论改造出的药物,而后续的药理研究表明,它其实是个前药,在体内通过形成磷酸化活性物质发挥作用。如果藤田采用分子水平模型而非细胞水平模型,那么将会与此化合物失之交臂。正是这种曲折历程,才让药物化学这门学科总是神秘与惊喜并存,令无数药化工作者流连忘返。
[1]Brinkmann V.FTY720(fingolimod)in Multiple Sclerosis:therapeutic effects in the immune and the central nervous system[J].Br J Pharmacol,2009,158(5):1173-1182.
[2]Miron VE,Schubart A,Antel JP.Central nervous system-directed effects of FTY720(fingolimod)[J].J Neurol Sci,2008,274(1-2):13-17.
[3]Ebers GC.A full genome search in multiple sclerosis[J]. Nat Genet,1996,13(4):472-476.
[4]Deogracias R,Yazdani M,Dekkers MP,et al.Fingolimod,a sphingosine-1 phosphate receptor modulator,increases BDNF levels and improves symptoms of a mouse model of Rett syndrome[J].Proc Nati Acad Scis,2012,109(35):14230-14235.
[5]Chiba K,Adachi K.Discovery of fingolimod,the sphingosine 1-phosphate receptor modulator and its application for the therapy of multiple sclerosis[J].Future Med Chem,2012,4(6):771-781.
[6]Fujita T,Matsumoto N,Uchida S,et al.Antibody against a novel,myriocin(ISP-I)-based immunosuppressant,FTY720[J].Bioorg Med Chem Lett,2000,10(4):337-339.
[7]St-Jacques M.Elucidation of structure and stereochemistry of myriocin.A novel antifungal antibiotic[J].J Org Chem,1973,38(7):1253-1260.
[8]Fujita T,Yoneta M,Hirose R,et al.Simple compounds,2-alkyl-2-amino-1,3-propanediols have potent immunosuppressive activity[J].Bioorg Med Chem Lett,1995,5(8):847-852.
[9]Suzuki S,Li XK,Enosawa S,et al.A new immunosuppressant,FTY720,induces bcl-2-associated apoptotic cell death in human lymphocytes[J].Immunology,1996,89(4):518-523.
[10]Hamamichi N,Fujita T,Matsuzaki T,et al.14 determination of the absolute configurations and total synthesis of new immunosuppressants,mycestericins D,E,F and G [J].Natural Organic Compounds Debate Abstracts(Japan),1995,37:79-84.
[11]Fujita T,Hirose R,Yoneta M,et al.Potent immunosuppressants,2-alkyl-2-aminopropane-1,3-diols[J]. J Med Chem,1996,39(22):4451-4459.
[12]Kiuchi M,Adachi K,Kohara T,et al.Synthesis and immunosuppressive activity of 2-substituted 2-aminopropane-1,3-diols and 2-aminoethanols[J].J Med Chem,2000,43(15):2946-2961.
[13]Sanchez T,Hla T.Structural and functional characteristics of S1P receptors[J].J Cell Biochem,2004,92(5):913-922.
[14]Sanchez T,Estrada-Hemandez T,Paik JH,et al.Phosphorylation and action of the immunomodulator FTY720 inhibits vascular endothelial cell growth factor-induced vascular permeability[J].J Biol Chem,2003,278(47):47281-47290.
[15]Pitman MR,Woodcock JM,Lopez AF,et al.Molecular targets of FTY720(fingolimod)[J].Curr Mol Med,2012, 12(10):1207-1219.
[16]Pchejetski D,Bohler T,Brizuela L,et al.FTY720(fingolimod)sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1[J].Cancer Res,2010,70(21):8651-8661.
[17]Paugh SW,Payne SG,Barbour SE,et al.The immunosuppressant FTY720 is phosphorylated by sphingosine kinase type 2[J].FEBS Lett,2003,554(1-2):189-193.
[18]郭宗儒.从天然产物到免疫调节药物芬戈莫德[J].药学学报,2014,49(1):148-150.
A research and development overview of Fingolimod
NIU Jun-xing,HU Li-hong
(Shanghai Research Center for Modernization of Traditional Chinese Medicine,Shanghai Institute of Materia Medica,Chinese Academy of Sciences,Shanghai 201203,China)
Fingolimod(FTY720),a novel immunomodulator agent,is developed from ISP-I,which is a secondary metabolites of fungi.It was approved by FDA in 2012 for the treatment of autoimmune disease multiple sclerosis.Unlike the traditional immunosuppressive agents,fingolimod exerts immunosuppressive and immunoregulatory functions mainly through interaction with shhingosine-1-phosphate(S1P)receptors on the cell surface without affecting activation and proliferation of lymphocyhtes.This article briefly reviewed the development process of fingolimod,elaborate the structural optimization and transformation,in order to provide experience for chemical rsearch.
Finglimod;ISP-I;Multiple Sclerosis;Immunomodulator
R979.1
A
2095-5375(2015)12-0683-006
国家自然科学基金面上项目(No.81473110)
猜你喜欢
中学化学(2022年5期)2022-06-17 16:51:48
中学生数理化·中考版(2021年12期)2021-12-31 03:24:40
中学生数理化·中考版(2021年11期)2021-12-06 07:29:16
高中数理化(2020年1期)2020-02-29 02:21:18
四川师范大学学报(自然科学版)(2018年2期)2018-04-28 02:21:08
中学化学(2017年6期)2017-10-16 17:22:41
郑州大学学报(理学版)(2014年2期)2014-03-01 04:20:55
昌吉学院学报(2013年1期)2013-12-08 07:36:22
