
口蹄疫病毒A/GDMM/CHA/2013株结构蛋白VP1 H-2d限制性CTL表位预测
2015-11-05 11:09尚延丽冯霞靳野张晓霞张柳马军武
安徽农业科学 2015年16期
尚延丽,冯霞,靳野,张晓霞,张柳,马军武
(中国农业科学院兰州兽医研究所家畜疫病病原生物学国家重点实验室/国家口蹄疫参考实验室,甘肃兰州730046)
口蹄疫(Foot-and-mouth disease,FMD)是由口蹄疫病毒引发的一种急性、烈性、高度接触性传染病,其严重危害畜牧业的发展和国家的经济贸易。在我国,口蹄疫的防制主要依赖于以强制免疫为主的综合免疫措施,但目前使用的灭活疫苗[1]存在着许多缺点,研究、开发安全、高效的新型疫苗[2-3]是大势所趋,而表位肽疫苗正是其研究热点之一。表位肽疫苗是指同时含有多个目标抗原表位和相应的辅助性表位的一种新型疫苗。抗原表位是指抗原分子表面上有特殊结构与免疫活性的化学基团,它代表抗原分子的一个免疫活性区,是能够刺激机体产生抗体或致敏淋巴细胞并且能够被其识别的部位。免疫细胞不是对完整的抗原分子进行识别,而是仅识别抗原肽上的表位。抗原表位按照与不同的抗原受体细胞结合,可分为B细胞抗原表位与 T细胞抗原表位,而T细胞表位可分为CTL表位和辅助性Th表位。
传统的表位筛选方法(如合成肽库法、步移重叠多肽法、酸洗脱法等)工作量大、试验成本高、试验周期长,也可能遗漏一些细胞表位。目前,细胞表位筛选方法主要是采用计算机预测软件预测与实验法鉴定相结合来筛选细胞表位。计算机表位预测是在已知抗原蛋白结构的基础上或者已知氨基酸序列的基础上根据氨基酸的理化数据特质或者肽片段与MHC分子结合、TAP转运过程、蛋白酶体裂解等抗原递呈过程的规律,将它们编入计算机的算法中,然后用计算机软件来预测可能含有表位的区域。笔者通过多种计算机表位预测软件预测口蹄疫病毒A/GDMM/CHA/2013株结构蛋白VP1上可能的CTL T细胞表位,综合分析各软件的预测结果,筛选出最有可能T细胞表位的候选表位,旨在为鉴定口蹄疫病毒CTL细胞表位、研发表位肽疫苗提供数据和基础。
1 材料与方法
1.1 FMDV A/GDMM/CHA/2013株结构蛋白 VP1的核苷酸和氨基酸序列 从GenBank中查询到FMDV A/GDMM/2013株结构蛋白VP1的序列,并与国内外口蹄疫参考毒株进行比较分析。
1.2 CTL T细胞表位预测 应用Syfpeithi、IEDB、NetMHC-3.2、IMTECH和BIMAS等多种生物信息学软件预测FMDV A/GDMM/2013株结构蛋白VP1上的T细胞表位(表1)。将FMDV A/GDMM/2013 VP1的氨基酸序列输入各个网站,分别预测了其H-2Dd、H-2Kd、H-2Ld限制性T细胞表位。候选表位氨基酸长度参数为9 mer。选取在各预测软件上分值在前10位,并且在其他软件前10位重复率高的肽段做为候选表位。

表1 试验所用的CTL表位预测软件
1.3 候选表位稳定性预测 使用在线分析软件ProtParam,分析候选表位的理化性质及其稳定性。
2 结果与分析
2.1 VP1基因序列分析 FMDV A/GDMM/2013株结构蛋白VP1基因(KF450794.1)全长636个核苷酸,编码212个氨基酸(图1~2)。通过GenBank同源性比对发现,FMDV A/GDMM/CHA/2013株结构蛋白 VP1基因与A/HuBWH/CHA/2009 VP1 protein(VP1)gene(JF792355.1)同源性为91%,与 A/VIT/11/2004 VP1(1D)gene(HQ116363.1)、A/TAI/7/2003 VP1(1D)gene(HQ116312.1)、/MAY/3/2003 VP1(1D)gene(HQ116297.1)同源性为94%。
2.2 VP1蛋白CTL细胞表位预测 将FMDV A/GDMM//CHA/2013结构蛋白VP1蛋白序列输入到预测软件,选好MHC的类型及候选表位的长度。每个软件均选取排名的前10位,然后在每个软件、每种MHC类型的前10中交叉比较选择排名靠前且重复率高的10个肽段为候选CTL表位(表2~3)。
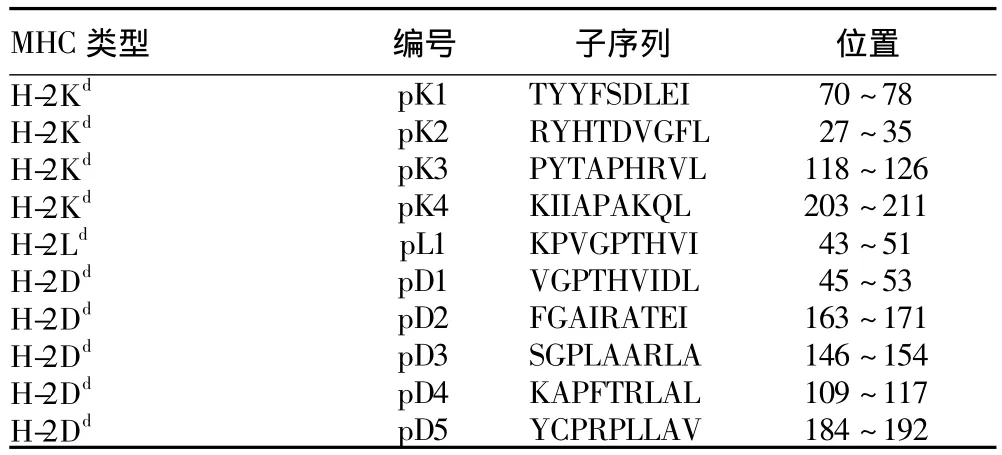
表2 预测软件预测的VP1蛋白的CTL表位氨基酸序列
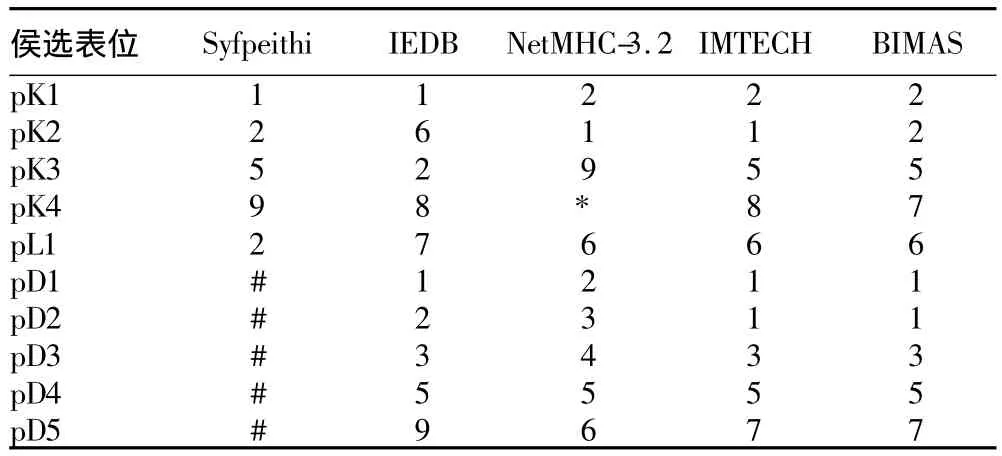
表3 筛选出的候选表位在各个软件中的排名
2.3 候选表位稳定性预测 采用在线分析软件ProtParam,分析筛选出的10条候选表位。由表4可知,pK1、pK4、pD2、pD4和pD5的不稳定指数分别为44.00、68.57、42.26、51.69和43.31。这5个表位不稳定指数较高,稳定性差,可能不是CTL表位。

表4 ProtParam分析结果
3 讨论与结论
CTL细胞表面的TCR能够识别细胞表面展示出的MHC-I-抗原肽(CTL细胞表位)复合物,并与之结合,然后杀伤靶细胞。CTL在抗肿瘤和抗病毒感染的免疫反应中发挥着重要的作用。当内源性抗原通过MHCⅠ类分子展示于细胞表面后,被CD8T细胞受体识别,在共刺激因子作用下CD8T被活化、增殖产生细胞毒T细胞(CTL)。CTL具有强大的杀伤靶细胞的能力。CTL通过杀伤介质(Perforin、Granayme、Fasl、TRAIL、TNF、IFN-γ 等)来杀伤靶细胞。CTL 在发挥效应时(识别靶细胞和攻击靶细胞)受MHCⅠ类分子的限制,即CTL只杀伤自身MHC递呈抗原的靶细胞。由于抗体只能直接结合体液中的抗原,不能通过细胞膜进入细胞内杀伤病原,因此细胞免疫对清除病毒和胞内寄生菌十分重要,CTL通过杀伤靶细胞破坏胞内病原赖以生存的环境,在抗体和吞噬细胞的配合下将病原彻底清除。MHC-Ⅰ类分子仅识别和结合内源性抗原表位,而CTL发挥效应时受MHC-Ⅰ的限制,因此仅由MHC-Ⅰ类分子转运的抗原表位才能够诱发CTL效应,这类抗原表位就是CTL表位。
研究表明,自然宿主抗口蹄疫病毒感染的能力与高水平的中和抗体水平有重要相关性。此外,中和抗体的产生离不开Th细胞的辅助作用,因此以前对口蹄疫病毒抗原表位的研究主要是针对B细胞表位和Th细胞表位。但是,有研究表明细胞免疫在FMDV免疫应答中也扮演着非常重要的角色[4-7]。因此,研究口蹄疫病毒的CTL表位具有重要意义,进一步充实了口蹄疫病毒表位数据库资料,也为深入了解细胞介导的抗口蹄疫病毒免疫铺垫了基础。
该研究的口蹄疫病毒A/GDMM/CHA/2013株是2013年从我国广东省茂名市分离的猪源毒,由国家参考实验室保存。该毒株属于A型ASIA拓扑性(东南亚)-97毒株,其基因与A/HuBWH/CHA/2009(JF792355)同源性小于91.5%,其基因与我国历史毒株AF/72的同源性小于81.6%,而其基因与2004年越南毒株A/VIT/11/2004大于93.7%。这表明它同我国历史毒株AF/72无直接遗传衍化关系,考虑到时间因素,其与2009年流行毒株A/HuB/WH/09应该也无直接遗传衍化关系,很可能是由境外传入。初步推测,我国现有疫苗可能对该毒株保护力有限。因此,研究该毒株的细胞表位、发展相应疫苗具有重要意义。
该研究采用多种在线表位预测软件来预测CTL表位。其中,BIMAS[8]和 SYFPEITHI正是采用矩阵方法开发的。IEDB中,MHC-I类结合预测工具则是综合了ANNS(人工神经网络)[9-12]、SMM(稳定矩阵法)[13]、AMMPMBEC(肽段-MHC结合能力与稳定矩阵法)[14]、CombLib(组合库矩阵法)[15]、Consensus(共 识 表 位 预 测 方 法)[16]、NeMHC-pan[17-18]、PickPocket(结合槽选择法)[19]、NetMHCcons(肽段-MHC结合预测综合算法)[20]等多种肽段结合预测方法,而NetMHC-3.2则是结合了人工神经网络和权重矩阵2种方法。计算机表位预测在CTL抗原表位预测方面取得了很多成果。Gao等使用SYFPEITHI和GENETXY软件预测O型FMDV结构蛋白VP1上表位,然后将预测的候选表位同重组、表达SLA-2-(G4S)3-β2m蛋白复合体结合,分析候选表位同SLA-2-(G4S)3-β2m蛋白复合体的结合能力,结果表明表位26–34(RRQHTDVSF)和157-165(RTLPTSFNY)为FMDV CTL表位[21]。Barfoed等用SYFPEITHI和BIMAS软件预测候选表位,然后构建了可表达2C蛋白和预测出的候选表位的质粒的DNA疫苗来免疫小鼠,通过胞内细胞因子染色法来检测免疫后小鼠CD8+细胞IFN-γ的合成,结果表明FMDV C-S8c1株2C蛋白上的第63~71位(KYKDAKEWL)是H-2-Kd限制性CTL表位[22]。Liu等利用BIMAS软件预测FMDV AF/72结构蛋白VP1的H-2d限制性T细胞候选表位,并且化学合成这些候选表位,然后用表达FMDV AF/72结构蛋白VP1免疫小鼠,将分离的免疫后小鼠脾淋巴细胞与候选表位体外共培养,采用T细胞增殖试验和IFN-ELISPOT试验进行验证,结果表明pK1(AYHKGPFTRL)是H-2Kd限制性T细胞表位pD7(GFIMDRFVKI)是H-2Dd限制性T细胞表位[23]。Gao等使用MetMHCpan-2.0和GENETYX预软件预测的O型FMDV结构蛋白VP1上的T细胞候选表位化学合成后,用脂质体包裹来免疫小鼠,并进行T细胞增殖试验、流式细胞术、IFN-ELISA、CTL试验、豚鼠免疫保护力试验及组织病理学观察,结果表明肽段VP126–34(RRQHTDVSF)和VP157–165(RTLPTSFNY)是O型FMDV的CTL表位,并且用其免疫动物可在一定程度上抵御口蹄疫病毒的攻击[24]。
但是,计算机预测表位也有诸多局限性,比如预测蛋白构象及表位递呈过程等都是极其复杂的问题,需要累积大量的试验数据,并且理论预测和实际情况总有一定差距。这就需要将2种或者多种以上的预测方法结合起来,以提高表位预测的准确率。该研究使用了5种预测软件,结合多种算法,综合考虑各个软件的预测结果,最终选出10条候选表位。
CTL表位主要是由8~11个氨基酸残基构成的线性表位。CTL表位是由内源性抗原在胞浆加工产生。内源性抗原是在细胞内产生和加工的蛋白,主要是细胞自身产生的蛋白、感染病毒或者细菌的细胞内产生的非细胞自身的蛋白和肿瘤蛋白等。蛋白酶体处理经泛素化后的内源性抗原,将其裂解成8~11个左右氨基酸残基的肽段,再经过转运蛋白(TAP)的作用进入内质网,与内质网上的MHCⅠ类分子上的抗原结合槽结合形成MHCⅠ-抗原肽复合物。然后,通过高尔基体转运到细胞膜表面,同CD8+T细胞上的TCR识别与结合,从而诱发CTL效应。因此CTL表位要有一定的稳定性,才能保证在整个加工、结合、递呈和识别过程的顺利进行,起到诱发细胞免疫的效应。因此,采用ProtParam软件对候选表位进行稳定性分析。结果表明,pK1、pK4、pD2、pD4和pD5的不稳定指数较高,分别为 44.00、68.57、42.26、51.69和43.31。因此,这5个候选表位可能不是CTL表位。
动物机体是一个复杂的、受多因素影响的、不断变化着的环境。笔者对候选表位所进行预测是在体外根据已知的细胞表位的各种理化数据、氨基酸残基出现的频率等多种因素来进行分析,其稳定性则是根据各个氨基酸残基理化性质来分析。因此,预测分析的候选表位可能与实际情况存在差异,还需要进行体内外试验来鉴定。
该研究借助于计算机表位预测软件和蛋白质稳定性分析软件来预测和分析口蹄疫病毒A/GDMM/CHA/2013株结构蛋白VP1上可能存在的H-2d限制性T表位,结果表明有5条候选表位可能是CTL表位,包括H-2Kd限制性表位2条(第27-35、118-126位)、H-2Ld限制性表位1条(第43-51位),以及H-2Dd限制性表位2条(第45-53、146-154位)。当然,该研究预测的表位还需要体内外试验来验证。
[1]DOEL T R.FMD vaccines.[J].Virus Res,2003,91(1):81-99.
[2]BERGMANN I E,MALIRAT V,NEITZER T E,et al.Improvement of a serodiagnostic strategy for foot-and-mouth disease virus surveillance in cattle under systematic vaccination:a combined system of an indirect ELISA-3ABC with an enzyme-linked immunoelectrotransfer blot assay[J].Arch Virol,2000,145(3):473-489.
[3]DE DIEGO M,BROCCHI E,MACKAY D,et al.The non-structural polyprotein 3ABC of foot-and-mouth disease virus as a diagnostic antigen in ELISA to differentiate infected from vaccinated cattle[J].Arch Virol,1997,142(10):2021-2033.
[4]SANZ-PARRA A,JIMENEZ-CLAVERO M A,GARCIA-BRIONES M M,et al.Recombinant viruses expressing the foot-and-mouth disease virus capsid precursor polypeptide(P1)induce cellular but not humoral antiviral immunity and partial protection in pigs[J].Virology,1999,259(1):129-134.
[5]MAYR G A,CHINSANGARAM J,GRUBAN M J.Development of replication-defective adenovirus serotype 5 containing the capsid and 3C protease coding regions of foot-and-mouth disease virus as a vaccine candidate J].Virology,1999,263(2):496-506.
[6]SANZ-PARRA A,VAZQUEZ B,SOBRINO F,et al.Evidence of partial protection against foot-and-mouth disease in cattle immunized with a recombinant adenovirus vector expressing the precursor polypeptide(P1)of foot-and-mouth disease virus capsid proteins[J].J Gen Virol,1999,80(Pt3):671-679.
[7]MORAES M P,MAYR G A,MASON P W,et al.Early protection against homologous challenge after a single dose of replication-defective human adenovirus type 5 expressing capsid proteins of foot-and-mouth disease virus(FMDV)strain A24[J].Vaccine,2002,20(11/12):1631-1639.
[8]BHASIN M,LATA S,RRAGHAVA G P.Searching and mapping of T-cell epitopes,MHC binders,and TAP binders[J].Methods Mol Biol,2007,409:95-112.
[9]LUNDEGAARD C,LAMBERTH K,HARNDAHL M,et al.NetMHC-3.0:accurate web accessible predictions of human,mouse and monkey MHC class I affinities for peptides of length 8-11[J].Nucleic Acids Res,2008,36:509-512.
[10]LUNDEGAARD C,NIELSEN M,LUND O.The validity of predicted T-cell epitopes[J].Trends Biotechnol,2006,24(12):537-538.
[11]NIELSEN M,LUNDEGAARD C,BLICHER T,et al.Reliable prediction of T-cell epitopes using neural networks with novel sequence representations[J].Protein Sci,2003,12(5):1007-1017.
[12]BUUS S,LAUEMOLLER S L,WORNING P,et al.Sensitive quantitative predictions of peptide-MHC binding by a‘Query by Committee’artificial neural network approach[J].Tissue Antigens,2003,62(5):378-384.
[13]PETERS B,SETTE A.Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method[J].BMC Bioinformatics,2005,6:132.
[14]KIM Y,SIDNEY J,PINILLA C,et al.Derivation of an amino acid similarity matrix for peptide:MHC binding and its application as a Bayesian prior.[J].BMC Bioinformatics,2009,10:394.
[15]SIDNEY J,ASSARSSON E,MOORE C,et al.Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries[J].Immunome Res,2008,4:2.
[16]MOUTAFTSI M,PETERS B,PASQUETTO V,et al.A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus[J].Nat Biotechnol,2006,24(7):817-819.
[17]HOOF I,PETERS B,SIDNEY J,et al.NetMHCpan,a method for MHC class I binding prediction beyond humans[J].Immunogenetics,2009,61(1):1-13.
[18]NIELSEN M,LUNDEGAARD C,BLICHER T,et al.NetMHCpan,a method for quantitative predictions of peptide binding to any HLA-A and-B locus protein of known sequence[J].PLoS One,2007,2(8):796.
[19]ZHANG H,LUND O,NIELSEN M.The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities:application to MHC-peptide binding[J].Bioinformatics,2009,25(10):1293-1299.
[20]KAROSIENE E,LUNDEGAARD C,LUND O,et al.NetMHCcons:A consensus method for the major histocompatibility complex class I predictions[J].Immunogenetics,2012,64(3):177-186.
[21]GAO F S,FANG Q M,LI Y G,et al.Reconstruction of a swine SLA-I protein complex and determination of binding nonameric peptides derived from the foot-and-mouth disease virus[J].Vet Immunol Immunopathol,2006,113(3/4):328-338.
[22]BARFOED A M,RODRIGUEZ F,THERRIEN D,et al.DNA immunization with 2C FMDV non-structural protein reveals the presence of an immunodominant CD8+ ,CTL epitope for Balb/c mice[J].Antiviral Res,2006,72(3):178-189.
[23]LIU X S,WANG Y L,ZHANG Y G,et al.Identification of H-2d restricted T cell epitope of foot-and-mouth disease virus structural protein VP1[J].Virol J,2011(8):426.
[24]GAO F S,FENG L,ZHANG Q,et al.Immunogenicity of two FMDV nonameric peptides encapsulated in liposomes in mice and the protective efficacy in guinea pigs[J].PLoS One,2013,8(7):68658.
猜你喜欢
遵义医科大学学报(2023年4期)2023-05-05
今日畜牧兽医(2022年10期)2022-12-23
今日农业(2021年8期)2021-11-28
中学生数理化·中考版(2021年9期)2021-11-20
临床医药文献杂志(电子版)(2017年11期)2017-05-17
临床骨科杂志(2017年1期)2017-03-07
中国免疫学杂志(2017年1期)2017-01-17
小学生导刊(低年级)(2016年7期)2016-07-29
畜牧兽医学报(2015年3期)2015-07-05
医学研究杂志(2015年6期)2015-07-01
