
紫脲酸铵分光光度法测定复盐法水氯镁石脱水产品中氧化镁
2015-10-25 02:17胡湖生杨明德吴玉龙
计量学报 2015年5期
胡湖生, 杨明德, 吴玉龙
(清华大学核能与新能源技术研究院,北京 102201)
紫脲酸铵分光光度法测定复盐法水氯镁石脱水产品中氧化镁
胡湖生, 杨明德, 吴玉龙
(清华大学核能与新能源技术研究院,北京 102201)
对于水氯镁石的复盐(C6H5NH2·HCl·MgCl2·6H2O)法脱水产品中微量MgO含量的测定,采用了直接测定样品水溶残渣中镁含量而得MgO含量的方法代替传统的间接法,获得了更准确的结果。用紫脲酸铵分光光度法测定了水溶残渣用稀H2SO4溶解后溶液中微量镁,研究了金属镁离子与显色剂的络合比、溶液酸度、显色时间对显色的影响,以及盐酸苯胺阳离子、Fe3+、Zn2+、Ba2+、Ca2+、Ni2+、Co2+等离子对显色的干扰及其消除。并采用修正法消除了显色后残余显色剂对测定镁的影响。结果表明,在镁离子浓度0~2 mmol/L范围内,修正吸光度与镁离子浓度呈线性关系,摩尔吸光系数为2.45×102L/(mol·cm)。加标镁离子的测定结果证明该法具有较高的精密度和准确度(加标回收率近100%),适合于水氯镁石脱水产品中总MgO含量的准确测定。
计量学;水氯镁石;紫脲酸铵;分光光度法;氧化镁;脱水产物
1 引 言
盐湖里含有巨大的镁资源,将水氯镁石脱水制取无水氯化镁进而电解制取金属镁是利用盐湖镁资源的一条重要途径。水氯镁石脱水制取无水氯化镁时很容易水解成碱式氯化镁进而转化为氧化镁[1~3],即使在用苯胺盐酸盐(C6H5NH2·HCl)作脱水剂的复盐法脱水工艺中在某些条件下也难免有少量氯化镁水解[4]。工业上对脱水产品中氧化镁采用了EDTA滴定容量差减法,即分别测定固体样品的醇溶、酸溶后液相中的镁含量,计算出氯化镁含量和总镁含量,利用差减法间接求得氧化镁的含量。此法用于测定样品中微量氧化镁时,会产生很大的误差,但如果直接测定脱水产品样品水溶渣中镁含量、再计算出氧化镁含量,可以避免产生上述问题,得到很精密的结果。测定水溶残渣中微量镁,用EDTA滴定法容易发生过量,原子吸收光谱法[5~8]设备昂贵,而用分光光度法正适于这种微量分析,且设备便宜。文献报道了偶氮氯膦I分光光度法[9]、二甲酚橙分光光度法[10]、酸性铬兰K分光光度法[11]、4-(2-吡啶基)间苯二酚[12]、邻甲酚酞络合剂[13]、羟基茜草素[14]、PA.FPNS[15]、茜素氨羧络合剂(AC)[16]、溴代邻苯三酚红[17]测定微量镁,但用显色敏锐的紫脲酸铵作显色剂的分光光度法测定微量镁还未见报道。
本文研究了紫脲酸铵(MX)分光光度法测定水溶液中镁离子的影响因素,并根据修正光度理论[18],消除了显色后残余显色剂本身颜色对测定的影响,使修正吸光度与镁离子浓度符合比耳定律,大大提高了分析灵敏度。进而测定了脱水产品中水溶渣中镁含量,再计算出MgO含量,结果令人满意。
2 实验部分
2.1仪器与试剂
仪器:(1)SPECORD S600型紫外分光光度计,由计算机通过WinASPECT软件控制;(2)pHS-25C型数字pH计。
试剂:(1)紫脲酸铵(分析纯)水溶液(3.48 mmol/L;(2)NH3·H2O-NH4Cl缓冲溶液([NH3· H2O]=7.83 mol/L,[NH4Cl]=1.25 mol/L,pH= 10.5);(3)EGTA(乙二醇二乙醚二胺四乙酸)溶液(2.6 mmol/L);(4)2.5%氨水;(5)0.05 mol/L H2SO4;(6)镁标准溶液0.05 mol/L。
2.2实验方法
2.2.1显色液制备及其吸光度测定
在25 mL比色管中,加镁标准溶液若干mL,加入紫脲酸铵(MX)水溶液5 mL、EGTA溶液3 mL、NH3·H2O-NH4Cl缓冲溶液(pH=10.5)5 mL,用去离子水定容。以水作参比,用1 cm比色皿,在350~700 nm范围内吸光度扫描记录光谱曲线。在纯MX吸光度曲线上查出波长470 nm和520 nm处的吸光度A1和A2,在Mg-MX络合物吸光度曲线上查出波长470 nm和520 nm处的吸光度A3和A4,计算修正吸光度:Ac=A3-kA4(其中k=A2/A1,只决定于纯MX吸光度曲线形状)。若作标准曲线,则改变25 mL系列比色管中镁离子浓度,分别测定修正吸光度,作Ac~C(镁离子浓度)曲线。
2.2.2样品测定
准确称取脱水产品样品0.200 0 g左右,置于50 mL锥形瓶中,加入20 mL去离子水,盖上磨口塞后磁力搅拌1 h充分溶解样品,用滤纸过滤,用10 mL去离子水洗涤锥形瓶2~3次,洗液也加入到滤纸上过滤,弃去滤液。滤纸上残渣用滴管吸取去离子水淋洗数次,至耗50 mL去离子水时止,丢弃洗液。用滴管吸取煮沸的5%硫酸热溶解滤纸上的沉淀(至耗去酸液约20 mL),再用洗瓶冲洗滤纸(至耗去离子水约20 mL),将洗水与酸溶解液合并,定容50 mL,取5~10 mL于25 mL比色管(其余操作同2.2.1),测定修正吸光度,用标准曲线法求出样品镁含量。
3 实验结果与讨论
3.1吸收光谱
在可见光范围内,纯紫脲酸铵(MX)溶液呈紫红色,而Mg-MX络合物呈黄色。在187~1 000 nm范围内进行样品扫描,取与测定有关的波段,得图1。可见,Mg-MX络合物最大吸收波长为470 nm,比纯MX的最大吸收波长520 nm紫移了50 nm,对比度不太大,故显色液中残余显色剂紫脲酸铵的吸光对Mg-MX络合物的测定有影响,在求络合物真实吸光度时需要对表观吸光度进行修正以消除显色剂的影响。
3.2显色剂用量
在系列25 mL比色管中,各加1 mL 0.05 mol/L MgCl2的条件下,当加MX小于2 mL时,随着MX的加入量增大,Mg-MX络合物在470 nm处最大吸光度缓慢增大;当加MX大于2 mL时,在470 nm处最大吸光度急剧增大,见图2。由图2可以看出,出现显著峰形的显色剂用量是2 mL以上,但一般吸光度不宜大于1,故最佳MX用量是5 mL。
3.3溶液pH影响
在系列25 mL比色管中加入5mL MX溶液、2 mL MgCl2溶液,用2.5%氨水和5%H2SO4调节酸度,测定470 nm处修正吸光度AC,得到图3。
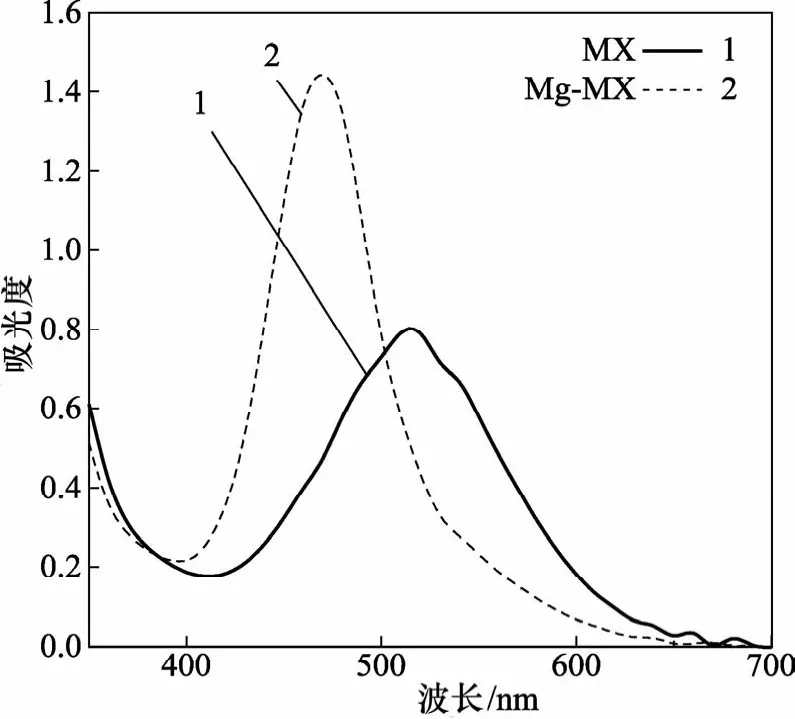
图1 纯MX与MX-Mg络合物吸收光谱图
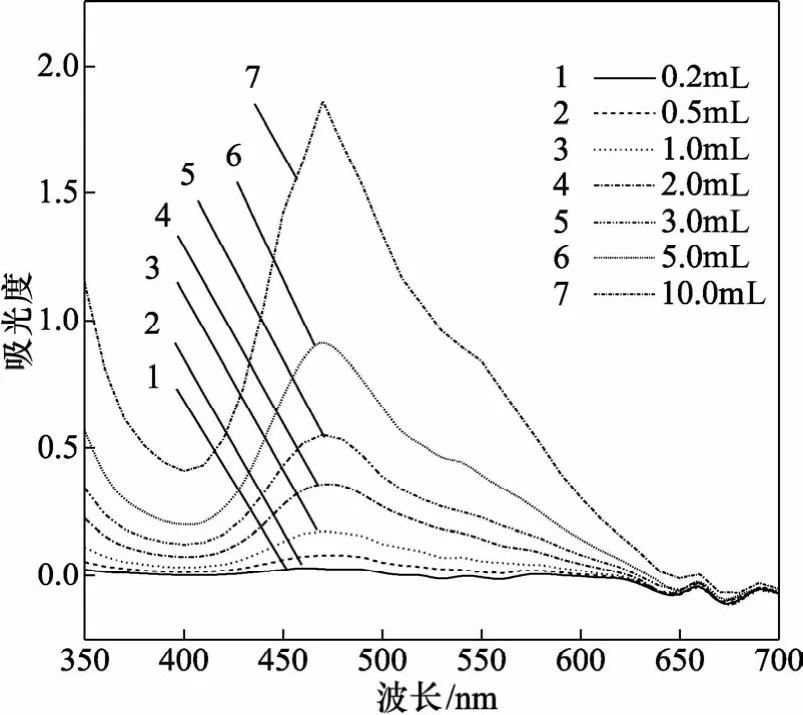
图2 MX用量的影响
由图可见,最佳pH值为10.5。因此必须用pH=10.5的氨水-氯化铵缓冲溶液控制比色溶液的酸度。
3.4显色时间与稳定性
显色后修正吸光度AC随时间变化见图4。由图可见,测定吸光度应在显色后20~40 min进行为宜。而且在对一系列显色溶液进行吸光度测定时采用测定顺序与加显色剂的先后顺序一致,可减少褪色引起的微量误差。
3.5Mg-MX络合物的络合比
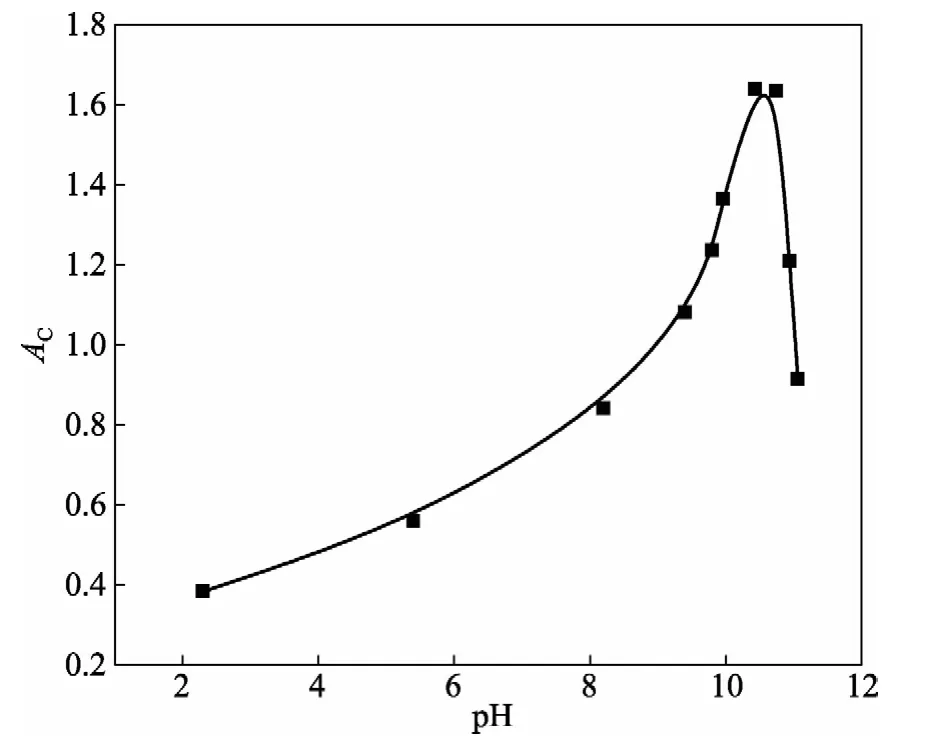
图3 溶液pH的影响
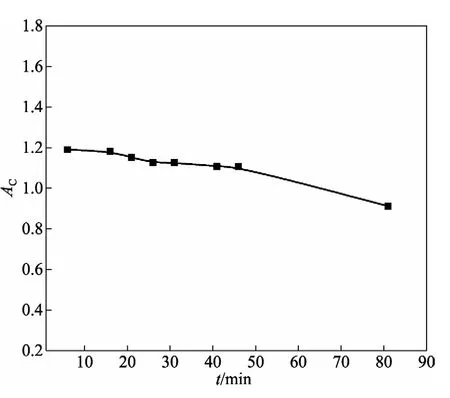
图4 Mg-MX络合物显色稳定性
采用摩尔比连续变化法求络合比。在系列25 mL比色管中,当0.003 45 mol/L的MX的剂量从2 mL逐渐减小至0.2 mL,同时0.05 mol/L的MgCl2的剂量从0.2 mL逐渐增大至2 mL时,溶液的吸光度变化见图5,图中ANC为470 nm处未修正吸光度。由图可见,470 nm处扣除试剂空白的吸光度随络合比变化曲线与修正吸光度随络合比变化曲线,均在[Mg2+]/[MX]=12处有最大值,说明Mg-MX络合比为12∶1。但未修正吸光度曲线波动太大,有时甚至出现负值,说明残余显色剂对络合物的测定有显著影响。而修正吸光度曲线比较稳定平滑,不会出现负值。
值得注意的是,若将上述实验条件中MgCl2浓度换成0.02和0.005 mol/L(其它条件不变)分别进行实验,则得到Mg-MX络合比分别为5.8∶1和1.5∶1。因此Mg-MX络合比不是一个值,而是随MgCl2浓度变化而变化的多个值。

图5 Mg-MX络合比
3.6标准曲线
在系列25 mL比色管中,各加3.45 mmol/L MX 2 mL的条件下,分别加入0.05 mol/L MgCl2的标准液0.0,0.2,0.5,1.0,2.0,4.0 mL,或0.05 mol/L MgCl2的加入量从0.2 mL逐渐增加至20 mL。在470 nm处测得扣除试剂空白的吸光度和修正吸光度,作AC~C(镁离子浓度)标准曲线。结果表明,这两种吸光度均随MgCl2浓度增加而增大,但修正吸光度曲线在低浓度范围内的线性相关系数(R2)比未进行修正的高得多。
未修正吸光度(以显色剂空白作参比)与镁离子浓度之间线性关系较差,如在镁浓度0~0.4 mmol/L范围内回归方程为ANC=0.293 48C+ 0.010 16,R2=0.974,C单位为mmol/L,见图6;而修正吸光度与镁离子浓度呈良好线性关系,即使在较大的浓度范围如在镁浓度0~2.0 mmol/L内,也较好地符合比耳定律,其标准曲线方程为:AC=0.244 75C+0.012 69,R2=0.999,C单位mmol/L,摩尔吸光系数为2.45×102L/(mol· cm),见图7。因此测定样品时应选择修正的标准曲线作为工作曲线。
3.7干扰离子的影响及其消除

图6 未修正的标准曲线

图7 修正的标准曲线
本文进行了离子干扰实验。对于25 mL溶液中含Mg2+100 μmol、各种干扰离子也分别为100 μmol的显色水溶液样品进行光谱扫描,结果见图8(a)与(b)。由图8(a)可见,100 μmol的Fe3+、Zn2+、Ba2+对镁离子的测定几乎无影响(吸光度AC变化不大于10%);但由图8(b)可见,100 μmol的Ca2+、Ni2+、Co2+对测定有显著地干扰,与Mg2+吸收峰位置相比,Ca2+使峰红移40 nm;Ni2+和Co2+使峰分别紫移30 nm和36 nm;而盐酸苯胺没有使峰移动但削弱了Mg2+的吸光度,只有当C6H5NH2·H+小于10 μmol时才无影响。更详细地实验研究证明,在25 mL显色溶液中存在100 μmol Mg2+的条件下,各干扰离子引起吸光度变化不超过5%的数量上限是:Na+2000 μmol、K+500 μmol、Zn2+35 μmol、Ba2+25 μmol、C6H5NH2·H+10 μmol、Fe3+和Cu2+各为2.5 μmol;Ca2+0.75 μmol;Ni2+和Co2+各为0.25 μmol。
实验证明,用EGTA(乙二醇二乙醚二胺四乙酸)可有效掩蔽Ca2+、Ni2+、Co2+对测定的影响。对于100 μmol Mg2+的水溶液,加入3 mL EGTA(2.6 mmol/L)溶液可掩蔽Ca2+2.5 μmol、Ni2+2.5 μmol、Co2+3.2 μmol。样品中盐酸苯胺阳离子(C6H5NH2·H+)可在样品制作时将其水溶、过滤撇去。此外,在pH=10条件下用三乙醇胺掩蔽Fe3+、Zn2+、Ba2+并无效果,反而对Mg2+显色稍有削弱作用,故不宜使用。

图8 干扰离子对吸光度曲线的影响
3.8精密度与加标回收率
对5 mL MX,1 mL 0.05 mol/L MgCl2溶液在470 nm下吸光度平行测定8次,得相对标准偏差为1.7%。
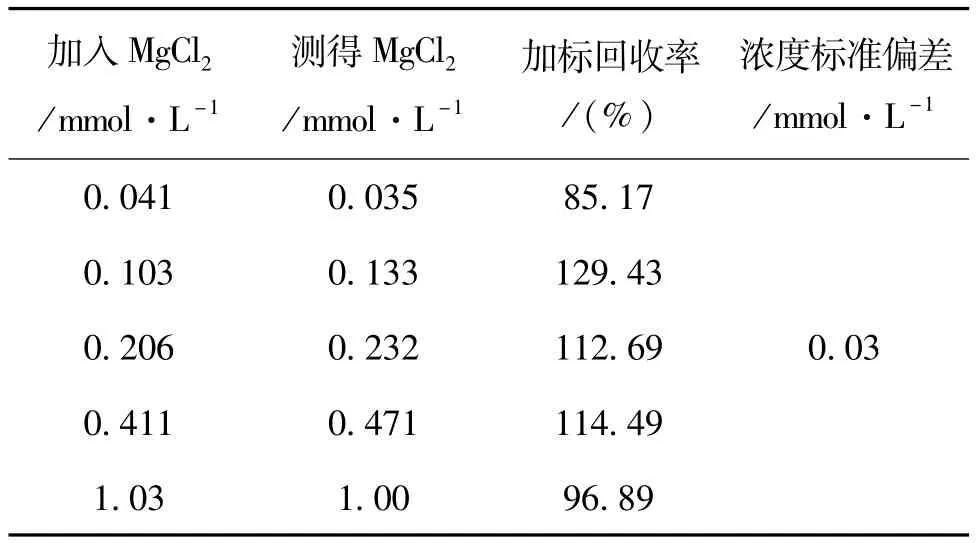
表1 加标回收率
由表1可见,在加标MgCl20.041~1.03 mmol/L范围内,加标回收率为85%~115%。
4 中试样品的测定
对于中试实验的同一炉4盘脱水产品,进行样品水溶、过滤,水溶渣洗涤后稀酸溶解,用分光光度法进行了测定,换算出水溶渣中氧化镁,结果见表2。测得结果与原子发射光谱法(ICP-AES)测得的结果很接近。
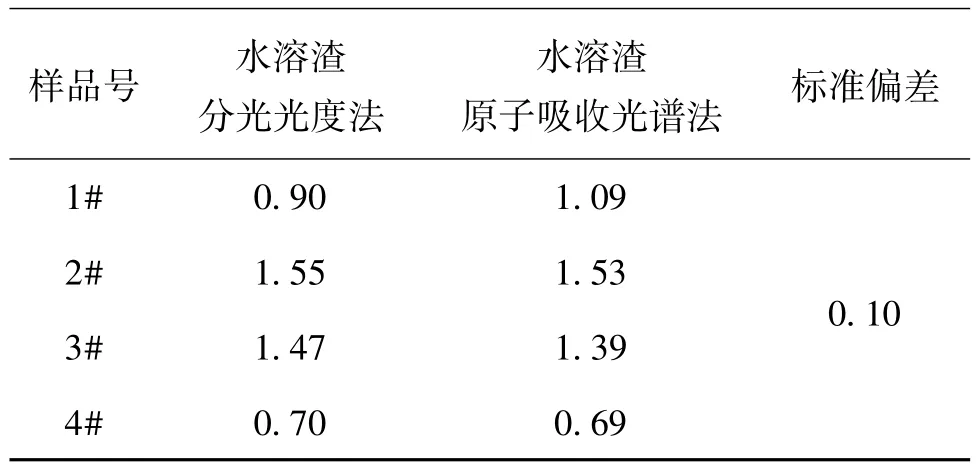
表2 中试脱水产品样品MgO分析结果(%)
5 结 论
用紫脲酸铵作显色剂的分光光度法测定水溶液中微量镁离子是可行的。溶液中共存离子如Fe3+、Zn2+、Ba2+对测定无影响;用EGTA可以掩蔽Ca2+、Ni2+、Co2+等离子对显色的干扰;盐酸苯胺阳离子可在制样时撇去。采用修正法可以消除显色后残余显色剂对测定镁的影响。实验结果表明,在镁离子浓度0~2mmol/L范围内,修正吸光度与浓度呈线性关系,摩尔吸光系数为2.45×102L/(mol·cm)。平行实验和加标镁离子的测定结果证明该法具有较高的精密度和准确度(加标回收率近100%),适合于水氯镁石脱水产品中MgO含量的准确测定。
[1] Sugimoto K,Dinnebier R E,Hanson J C.Structures of three dehydration products of bischofite from in situ synchrotron powder diffraction data(MgCl2·nH2O;n= 1,2,4)[J].Acta Crystallogr B:Struct Sci,2007,63(2):235-242.
[2] Kashani-NejadS,HarrisR.Characterizationof MgOHCl/MgO mixtures with infrared spectroscopy(IR)[C]//Conference on Magnesium Technology,Charlotte NC,2004.
[3] Huang Q Z,Lu G M,Wang J,et al.Thermal decomposition mechanisms of MgCl2·6H2O and MgCl2·H2O[J].J Anal Appl Pyrolysis,2011,91(1):159-164.
[4] 胡湖生,杨明德,吴玉龙.水氯镁石复盐法脱水工艺[J].有色金属(冶炼部分),2011,(7):17-21,38.
[5] 白钰,欧阳健明,白燕,等.火焰原子吸收光谱法同时测定尿中钙镁[J].光谱学与光谱分析,2004,24(8):1016-1019.
[6] GB/T 1513—2006矿石钙和镁含量的测定火焰原子吸收光谱法[S].
[7] 何建忠,吴胜,邵玉兰.火焰原子吸收光谱测定水质中钙、镁的方法改进[J].中国卫生检验杂志,2008,8(9):1903-1904.
[8] 孙莹莹,石榴花,孔可可.火焰原子吸收光谱法测定铝合金中镁[J].现代化工,2011,31(S1):432-433.
[9] 陈郁,马香娟.偶氮氯膦I分光光度法测定铝合金中髙含量镁[J].中国计量学院学报,2002,13(4):311 -314.
[10] 周正义,印天寿.二甲酚橙光度法快速测定植株钙、镁[J].安徽农业技术学院学报,1994,8(3):44 -52.
[11] 吕俐宾.酸性铬兰K分光光度法测定镁[J].贵州农业科学,2001,29(5):19-20.
[12] GómezE,EstelaJM,CerdàV.Simultaneous spectrophotometricdeterminationofcalciumand magnesium in water[J].Anal Chim Acta,1991,249:513-518.
[13] TesfaldetZO,StadenJFV,StefanRI. Spectrophotometricdeterminationofmagnesiumin pharmaceutical preparations by cost-effective sequential injection analysis[J].Talanta,2004,64(15):981-988.
[14] Idriss K A,Sedaira H,Ahmed H M.An insight into the solution equilibria of magnesium(II)with purpurin and spectrophotometric determination of magnesium[J]. Talanta,2001,54(2):369-375.
[15] Ma H M,Huang Y X,Liang S C.PA.FPNS:its synthesis and use in spectrophotometric determination of magnesium[J].Talanta,1996,43(1):21-26.
[16] Idriss K A,Sedaira H,Ahmed S S.Determination of strontium and simultaneous determination of strontium oxide,magnesium oxide and calcium oxide content of Portland cement by derivative ratio spectrophotometry[J].Talanta,2009,78(1):81-87.
[17] Benamor M,Aguerssif N.Simultaneous determination of calcium and magnesium by derivative spectrophotometry in pharmaceutical products[J].Spectrochim Acta Part A,2008,69(2):676-681.
[18] 章鹏飞,郜洪文.β修正一紫脲酸铵光度法测定铜[J].冶金分析,1995,15(5):28-31.
Murexide Spectrophotometric Determination of Magnesium Oxide in Dehydration Products of Bischofite from Hydrated Double Salt Method
HU Hu-sheng, YANG Ming-de, WU Yu-long
(Institute of Nuclear and New Energy Technology,Tsinghua University,Beijing 102201,China)
For determination of MgO in dehydration products of hydrated double salt(C6H5NH2·HCl·MgCl2·6H2O),that direct measurement of the MgO in the water-insolvable residue of dehydration product samples was substituted for the conventional indirect method yielded more accurate results.The water-insolvable residue was resolved by boiling diluted H2SO4solution and the microgram amount magnesium in the solution was measured by spectrophotometry,using murexide as a color reagent.The effects such as pH of solution,complex ratios of magnesium to murexide,dosages of chromogenic reagent,reaction time on the absorbances of coloured solutions,and the tolerance limits of some interfering ions such as C6H5NH2·H+、Fe3+、Zn2+、Ba2+、Ca2+、Ni2+、Co2+and their masking reagents are investigated.The calibration curve is established under completely deduction of the absorbance of remnant colour reagent according to the correction spectrophotometric theory.In the concentration range of 0~2 mmol/L of Mg2+,the calibration curve is linear and molar absorptivity 2.45×102L/(mol·cm).The experimental results show that this method can give high precisions and accuracies(recoveries of standard addition were close to 100%),therefore it is suitable for analysis of the MgO in dehydration products.
metrology;bischofite;murexide;spectrophotometric;MgO;dehydration products
TB99
A
1000-1158(2015)05-0555-06
10.3969/j.issn.1000-1158.2015.05.23
2014-05-22;
2015-03-29
国家科技支撑计划重点项目(2008BAB35B05)
胡湖生(1965-),男,江西宁都人,高级工程师,硕士,研究方向为化工冶金、溶剂萃取技术。huhusheng123@tom.com
猜你喜欢
安徽化工(2022年4期)2022-08-02
山东冶金(2022年3期)2022-07-19
皮革制作与环保科技(2022年4期)2022-04-29
供水技术(2021年3期)2021-08-13
造纸化学品(2019年6期)2020-01-17
现代园艺(2017年21期)2018-01-03
广州化工(2016年6期)2016-09-05
云南中医学院学报(2014年5期)2014-07-31
化学分析计量(2013年5期)2013-03-11
云南中医学院学报(2012年3期)2012-07-31
