
CrCl3 对石油稠油O-H…X(X=N﹑O﹑S)氢键削弱的密度泛函理论研究
2015-10-25 03:11:14刘红飞
化工技术与开发 2015年8期
刘红飞
(宁夏大学物理电气信息学院,宁夏 银川 750021)
石油作为社会发展的主要能源之一,随着工业的发展和进步,常规石油储量急剧减少,石油危机成为制约社会发展的重要因素之一。为了保障社会的可持续发展,稠油成为了人们研究的重点[1-3]。稠油相对于常规石油而言,主要是由沥青质和胶质组成,分子量大,黏度大,不易开采和运输,但是其储量非常丰富,是常规原油的3 倍,组成多为含烷基支链和含杂原子的多环芳核和环烷芳核形成的三维缔合网络复杂结构,含有大量的O﹑N﹑S 杂原子,广泛分布在羟基﹑酯基﹑氨基﹑硫羟基等基团中,同稠油中富含的酸类化合物中羧基形成了O-H…N﹑O-H…O﹑O-H…S 等氢键和其它非常规氢键,O-H…N﹑O-H…O﹑O-H…S 的键强最大,造成内聚力增大,容易聚集,表现出黏度大的性质[4-7]。由于稠油组成比较复杂,用简单的分子吡啶中的N 原子﹑呋喃中的O 原子﹑噻吩中的S 原子作为氢键的质子受体,丙酸中的羧基上的O-H 基团作为供体,这样就构成O-H…N﹑O-H…O﹑O-H…S 氢键,以CrCl3作为催化剂去削弱氢键,运用密度泛函理论(DFT),对上述所选定的系统构建模型,并进行计算研究,解释其削弱氢键达到降低稠油黏度的机理。
1 计算方法
采用基于密度泛函理路(DFT)的B3LYP 方法,计算软件使用Gaussion03 程序[8]。在基组方面,Cr原子使用Lanl2dz 基组[9-10],C﹑H﹑O 使用6-31+G*基组,此方法在计算过渡金属的开壳层体系的电子结构的相关计算中,具有效率高,又能同时满足精度要求的优点。
2 结果及讨论
从键长看,同时对比图1 中添加CrCl3前后的几何结构图和表1 中的键长,可以看出加入CrCl3后,O14-H22…N3形成的氢键发生了明显的变化,其H22…N3的键长由原来的1.728A◦变为4.456A◦,N3和H22的作用距离明显增加,作用力显著减小。而同时O14-H22的键长由原来的1.01A◦变为0.989A◦,变化不大。因此从键长上看O14-H22…N3氢键大为削弱,N3向左偏转,O14-H22向右偏转。N3﹑H22﹑O14构成的键角也由以前的近直线的179.29°之间变为132.5°,H22…N3的作用力和O14-H22的作用力的合力也大为降低。O19-H20…O5形成的氢键键长发生了明显的变化,其H20…O5的键长由原来的1.956A◦变为3.917A◦,O5和H20之间的距离明显增加,作用力显著减小。而同时H20-O19的键长由原来的0.974A◦变为0.981A◦,距离变化不大,因此从键长上看O19-H20…O5氢键大为削弱。同时对比图1 中的b 和e 可以看出,加入CrCl3后,C4H4O 向左旋转导致O5向左旋转,CH3CH2COOH 中的H20-O19逆时针方向向右偏转,同时二者之间距离拉开。CH3CH2COOH 和C4H4O 的距离增加,作用力减小。O19-H20…S5形成的氢键发生了明显的变化,其H20…S5的键长由原来的2.588A◦变为4.411A◦,S5和H20的作用距离明显增加,作用力减小显著。而同时H20-O19的键长由原来的0.977A◦变为0.989A◦,距离变化不大,因此从键长上看O19-H20…S5氢键的键强大为削弱。加入CrCl3后,C4H4S 顺时针方向旋转,CH3CH2COOH 中的H20-O19相对于C4H4S 逆时针方向偏转,同时二者之间距离拉开。综合键长看O-H…N﹑O-H…O﹑O-H…S 键长明显增加,氢键得到削弱。
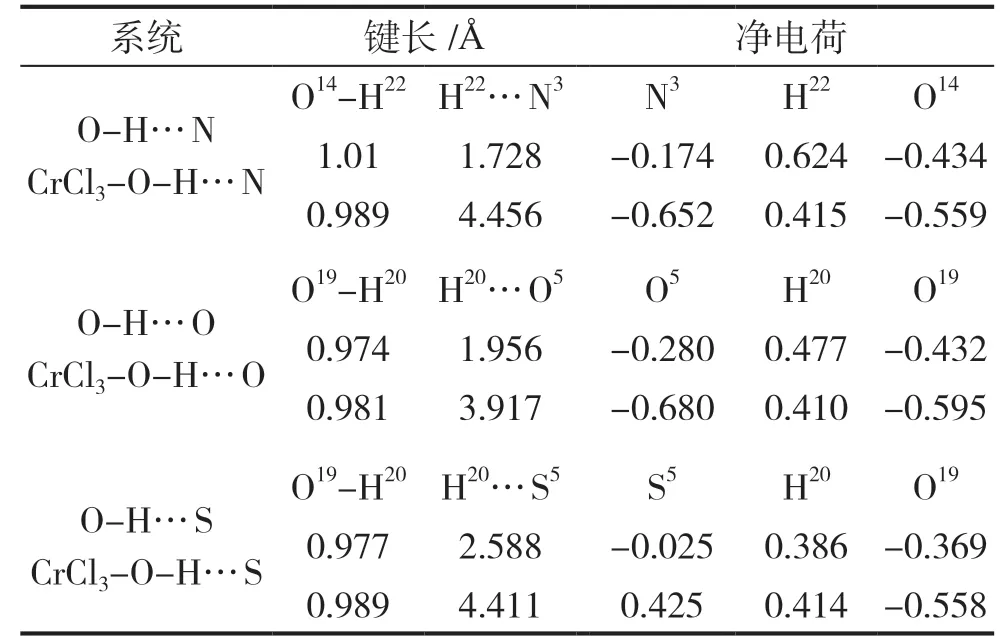
表1 CrCl3 对O-H…X(X=N、O、S)系统的键长和净电荷Table 1 The bond length and net charge of the CrCl3-O-H…X(X=N﹑O﹑S)
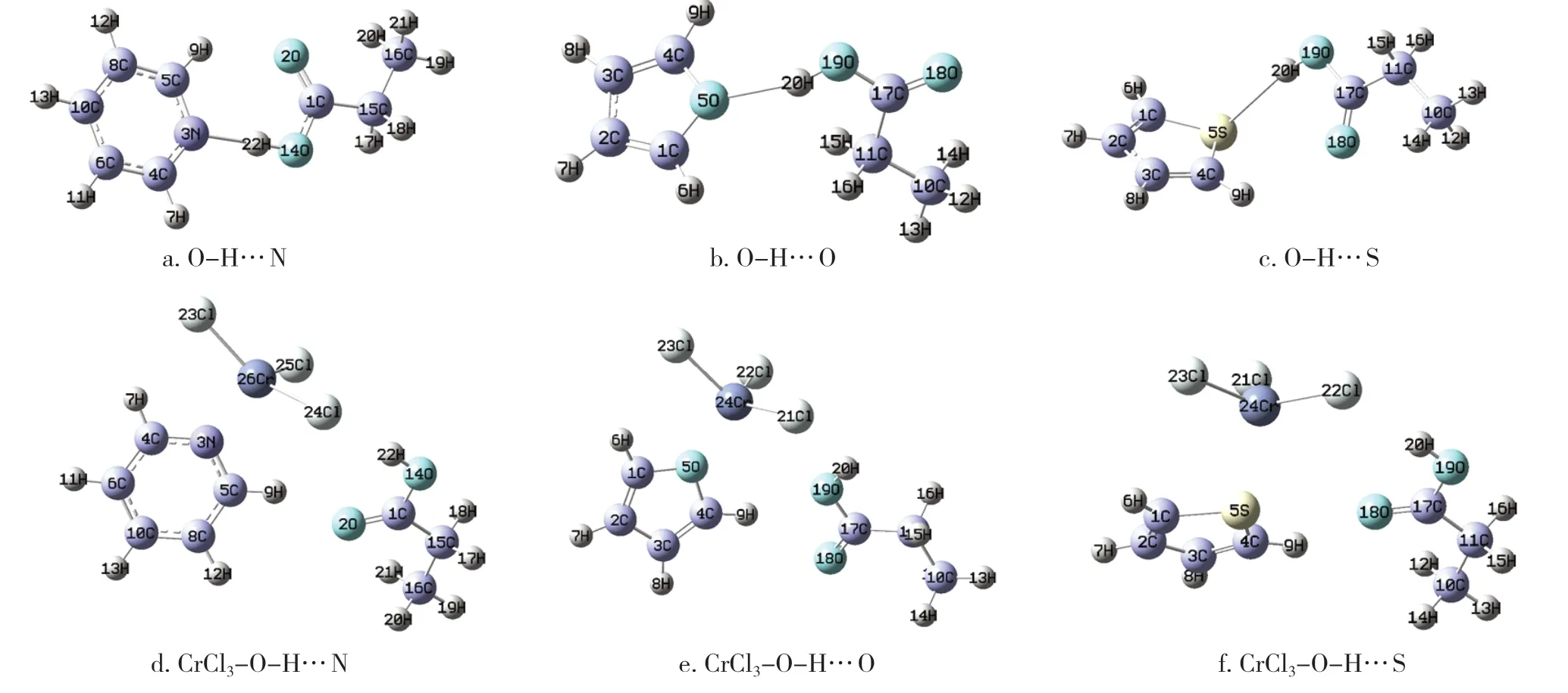
图1 CrCl3-O-H…X(X=N、O、S)的优化结构Figure 1 the optimizied structure of CrCl3-O-H…X(X=N﹑O﹑S)
从净电荷看,O14-H22…N3氢键中N3所带的电荷由-0.174 变为-0.652,电荷增加了0.478,H22所带的电荷由0.624 变为0.415,电荷降低了0.209,O14所带的电荷由-0.434 变为-0.559,电荷增加了0.124,虽然N3和O14电荷都增加了,但是氢键的强弱与键长和电荷都有关系,由于键长增加得更为大一些,因此这时键长的变化是主要因素,因而整体会表现出氢键削弱的状况。O19-H20…O5氢键中O5所带的电荷由-0.280 变为-0.680,增加了0.4,H20所带的电荷由0.477 变为0.410,降低了0.067,O19所带的电荷由-0.432 变为-0.595,增加了0.163。虽然O5和O19电荷都增加了,但是氢键的强弱与键长和电荷都有关系,由于键长增加得更大一些,因此这时键长的变化是主要因素,因而整体会表现出氢键削弱的状况。O19-H20…S5氢键中S5所带的电荷由-0.025 变为0.425,S 原子的电荷向周围的C4﹑C3﹑C2﹑C1转移,H20所带的电荷由0.386 变为0.414,O19所带的电荷由-0.369 变为-0.558,因此可知,在没有加入CrCl3之前,O19-H20…S5氢键的两端S5和O19都带的是负电荷,这样对H20产生相互吸引作用,使得氢键把CH3CH2COOH 和C4H4S 紧密地结合在一起,但是加入CrCl3之后,S5带正电荷,因此和H20产生排斥作用,同时将H20推向O19,而O19电荷由于增加到-0.558,也将H20向O19吸引靠近,使氢键趋于断裂。
从 能 量 上 看(表2),在O-H…X(X=O﹑N﹑S)氢 键 中,O-H…O 键 最 强,其 能 隙 最 高 为146.2kcal·mol-1,最不易被削弱,原因在于,一方面O 的电负性为3.44,大于N 和S 的电负性(3.04 和2.58)。对O-H…N 系统而言,系统的α 前线能隙为-71.7kcal·mol-1,β 前线能隙为-73.3kcal·mol-1,这2 个前线能隙小于没有加催化剂时的前线能隙-130.8kcal·mol-1。对O-H…O 系统而言,α-Homo能量为-170.1kcal·mol-1,β-Lumo 为-95.1kcal·mol-1,前线能隙为85kcal·mol-1,系 统 的β-Homo 能量为-74.9kcal·mol-1,β-Lumo 为-71.8kcal·mol-1,前线能隙为3.1kcal·mol-1,这2 个前线能隙小于没有加催化剂时的前线能隙146.2kcal·mol-1。O-H…S 系统α-Homo 能量为-170.1kcal·mol-1,β-Lumo为-98.1kcal·mol-1,前线能隙为72 kcal·mol-1,系统 的β-Homo 能 量 为-160.2kcal·mol-1,β-Lumo为-87.5kcal·mol-1,前 线 能 隙 为72.7kcal·mol-1,这2 个前线能隙小于没有加催化剂时的前线能隙136.1kcal·mol-1。对O-H…X(X=O﹑N﹑S)而言,加入CrCl3后,系统的能隙均有较大幅度降低,电子更容易发生跃迁,系统的稳定度降低,更加活跃,氢键明显弱化,达到了削弱目的。
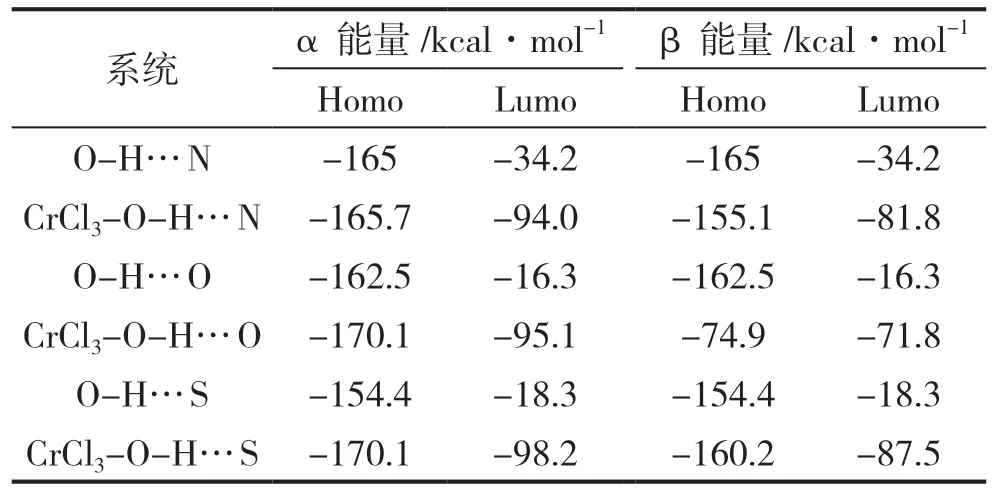
表2 CrCl3 对O-H…X(X=N、O、S)的系统能量Table 2 The energy of the CrCl3- O-H…X(X=N﹑O﹑S)
3 结论
计算表明,CrCl3的加入,使氢键键长明显增加,电荷发生转移,同时系统的前线能隙大幅降低,电子更容易发生跃迁,系统的稳定度降低,达到了削弱氢键的目的。这表明CrCl3具有较好的削弱氢键能力。
[1] 刘永建,钟立国,范洪富,等.稠油的水热裂解反应及其降黏机理[J].大庆石油学院学报,2002,26(3):95-98.
[2] 李美蓉,向浩,马济飞.特稠油乳化降黏机理研究[J].燃料化学学报,2006,34(2):175-178 .
[3] 刘永建,胡绍彬,闻守斌,等. 地质催化稠油水热裂解反应可行性研究[J].特种油气藏, 2007,14(5):84-87.
[4] Wilburn B E. Methacrylate pour point depressants and compositions: US, 4956111 [P]. 1991.
[5] Mishra M K, Raymond G. Pour point depressantsvia anionic polymerization of (meth) acrylicmonomers: US, 5834408[P].1998.
[6] Fischerj Terpolymer of ethylene ,vinyl acetate and is obutylene use ful as pour point depressants in crude oils: US,5256166[P]. 1993.
[7] Ritter Z. New polymers, mixtures thereof with poly(meth)acrylate esters and the use theeof for improving the cold fluidity of crude oil: US, 4906702[P]. 1990.
[8] Frisch M J, Trucks G W, Schlegel H.B, et al. Gaussian 03(revision B.05). Pittsburgh, PA: Gaussian,Inc, 2003.
[9] P J Hay, W R Wadt. Ab inito effective core potentials for molecular calcuations: potentials for K to Au including the outer most core orbitals[J]. J. Chem. Phys., 1985, 82(1): 290-310.
[10] P J Hay, W R Wadt. Ab inito effective core potentials for molecular calcuations: potentials for the transition metal atoms Sc to Hg[J]. J. Chem. Phys.,1985, 82(1): 270-283.
猜你喜欢
物理学报(2021年12期)2021-07-01 09:42:56
数学物理学报(2020年6期)2021-01-14 01:00:46
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
红领巾·探索(2017年9期)2017-10-26 22:23:41
中学生数理化·中考版(2016年10期)2016-12-22 18:31:56
武汉工程大学学报(2016年1期)2016-04-07 02:01:11
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
华南师范大学学报(自然科学版)(2013年1期)2013-10-27 10:51:51
