
冷冻水产品中乙萘酚残留的测定与降解规律
2015-10-16 07:19:12刘伶俐杨罗辉王冬武李晓玲
分析科学学报 2015年2期
刘伶俐, 洪 波*, 杨罗辉, 王冬武, 高 峰, 罗 玲, 刘 丽, 李晓玲
(1.湖南省水产科学研究所,湖南长沙 410153;2.长沙环境保护职业技术学院,湖南长沙 410004)
乙萘酚(2-Naphthol)是萘的2位氢被羟基取代后形成的化合物,是1-萘酚的异构体,主要用于制作防老剂乙萘酚、有机颜料及杀菌剂,同时作为防腐剂被广泛应用于食品行业,其污染载体涉及大气、水体以及食品。乙萘酚作为食品添加剂在使用量和使用途径上没有严格的控制和规范的管理。虽然水产品中暂未出现相关报道,但出于对水产品质量安全以及人体健康的考虑,建立乙萘酚在冷冻水产冻中的残留检测方法意义重大。
目前乙萘酚的测定方法主要有高效液相色谱[1,2]、高效液相色谱-串联质谱法[3]、气相色谱-质谱法[4]、直接荧光光度法[5],此外还有毛细管区带电泳法[6],而用于水产品中乙萘酚的检测方法未见报道。本文借鉴水果、蔬菜、水体的测定方法,拟建立一种准确、简便的检测方法来测定水产品中乙萘酚残留物。乙萘酚在食品当中残留时间长、降解速率慢。本实验期望通过测定不同添加浓度和储存时间的乙萘酚样品的残留量,揭示冷冻水产品中乙萘酚的自然降解规律。
1 实验部分
1.1 仪器与试剂
Waters e2695高效液相色谱仪(美国,Waters公司),配荧光检测器(FD);Q-101平行蒸发器(瑞士,Buchi公司);Z323K高速冷冻离心机(德国,Hemel公司);KQ5200DE超声波清洗器(昆山超声波仪器厂);C18柱(500 mg/3mL,美国Varian科技有限公司);LC-NH2固相萃取柱(500 mg/3mL,美国Varian科技有限公司)。
乙萘酚标准品(99%,国家标准物质研究中心)。取乙萘酚标准品配制成质量浓度为100 μg/mL的贮备液,再分别移取适量的贮备液配制成质量浓度分别为0.50、1.00、2.00、5.00、10.00 μg/mL的标准工作液。甲醇、乙腈、正己烷、乙酸乙酯(色谱纯,美国Tedia 公司);甲苯(色谱纯,天津科密欧化学试剂有限公司);酸化乙腈:在1 L乙腈中加入8 mL 50%HCl;乙腈-甲苯溶液:乙腈和甲苯按体积比3∶1混合。实验用水为超纯水(≥18.2 MΩ·cm)。
1.2 样品制备
取出冷冻水产品供试样品,在常温下自然解冻,取适量的样品制成细小、均匀的鱼糜样。
1.3 样品提取与净化
称取5.00 g(±0.02 g)的样品置于50 mL离心管中,加入15 mL酸化乙腈,匀浆30 s,35 ℃超声提取10 min,5 000 r/min离心10 min,将上清液转移至平行蒸发管中,重复提取1 次,上清液合并至蒸发管中,经40 ℃平行蒸发至近干,加入3 mL乙腈-甲苯溶液溶解残留物。
将蒸发管置于-60 ℃超低温冰箱中,冷冻处理15 min,取出后立即将液体全部转移至使用5 mL 乙腈、5 mL 乙腈-甲苯溶液活化的LC-NH2固相萃取柱,用4.00 mL乙腈-甲苯溶液洗脱,40 ℃氮气吹至干,用1.00 mL流动相溶解后,经0.22 μm微孔滤膜过滤,供高效液相色谱仪分析。
1.4 色谱条件
色谱柱:Inertil ODS-SP C18柱(250×4.6 mm,5 μm);流动相:乙腈∶水=50∶50(V/V);柱温:35 ℃;流速:0.80 mL/min;进样量:30 μL;检测波长:λex=250 nm,λem=410 nm。
2 结果与讨论
2.1 检测波长的确定
根据文献报道,扫描乙萘酚标准溶液的荧光光谱,结合样品基质的响应强度,确定方法的激发波长为激发波长λex=250 nm,发射波长λem=410 nm。
2.2 提取溶液与提取方式的选择
目前乙萘酚样品的处理方式主要是有机试剂浸提[7]、溴酸钾-罗丹明6G催化[8]等方式。实验以乙腈-水、乙腈及酸化乙腈做提取剂进行对比试验。在相同条件下,以乙腈-水配制成体积比分别为70∶30、50∶50、30∶70的提取液分次提取后,加入乙酸乙酯进行反萃取,离心后上下层之间有乳白色絮状物形成,影响提取效果,且乙酸乙酯的反萃取效率较低,回收率仅为20.3%;乙腈分次提取的效果明显好于乙腈-水,回收率在65.5%左右,但在提取如南美白对虾、鳗鲡等脂肪、蛋白质含量高的样品时易在提取液下层形成一定体积的油状物,从而对样品浓缩和定容造成影响;采用酸化乙腈较好地解决了蛋白质乳化影响,分次提取后浓缩的样品色谱图干扰峰极少,回收率较高,均在70.3%以上。实验选用酸化乙腈作为乙萘酚样品的提取溶剂。
2.3 净化方式的选择
乙萘酚样品的净化以固相萃取、液液萃取、分散液相微萃取等方式为主。实验分别对C18柱净化、冷冻去脂与LC-NH2固相萃取组合净化两种方式进行了比较。前一种方式在样品浓缩后分别加入3 mL甲醇和3 mL乙腈-甲苯溶液分次溶解残留物,经C18柱净化;后者直接采用乙腈-甲苯溶液溶解,冷冻去脂与LC-NH2固相萃取组合净化处理。结果显示:前者的分析图谱无干扰影响,但回收率始终低于65.1%;而后一种方式处理的各类样品的回收率均在70%以上,且样品在此色谱条件下分离效果更好,满足分析要求。因此确立酸化乙腈提取、冷冻去脂与LC-NH2固相萃取组合净化的方式。
2.4 标准工作曲线与方法检出限
按1.4节的仪器条件对系列标准工作液分别进样分析,以峰面积对浓度作图,绘制标准工作曲线,以3 倍信噪比(S/N)计算检出限。结果表明:乙萘酚的方法检出限为0.10 mg/kg,在0.50~10.00 μg/mL质量浓度范围内,其峰面积与质量浓度线性关系良好,相关系数R2=0.9998。
2.5 回收率与精密度
以草鱼、鳗鲡、南美白对虾的阴性样品添加0.50、1.00、5.00 mg/kg 3 个浓度的乙萘酚标准溶液,每个浓度做3个平行,按1.3节进行处理。计算出乙萘酚加标样品的平均回收率为74.8%~91.7%,相对标准偏差(RSD)为2.5%~9.4%,均满足实验室药物残留检测的要求,相关数据如表1所示。
2.6 实际样品的测定
在通过对标准物质和添加样品反复试验验证方法的准确性和可靠性的基础上,采用本方法对3 个批发市场随机抽取的20 个冷冻样品进行分析测定,结果均为阴性。
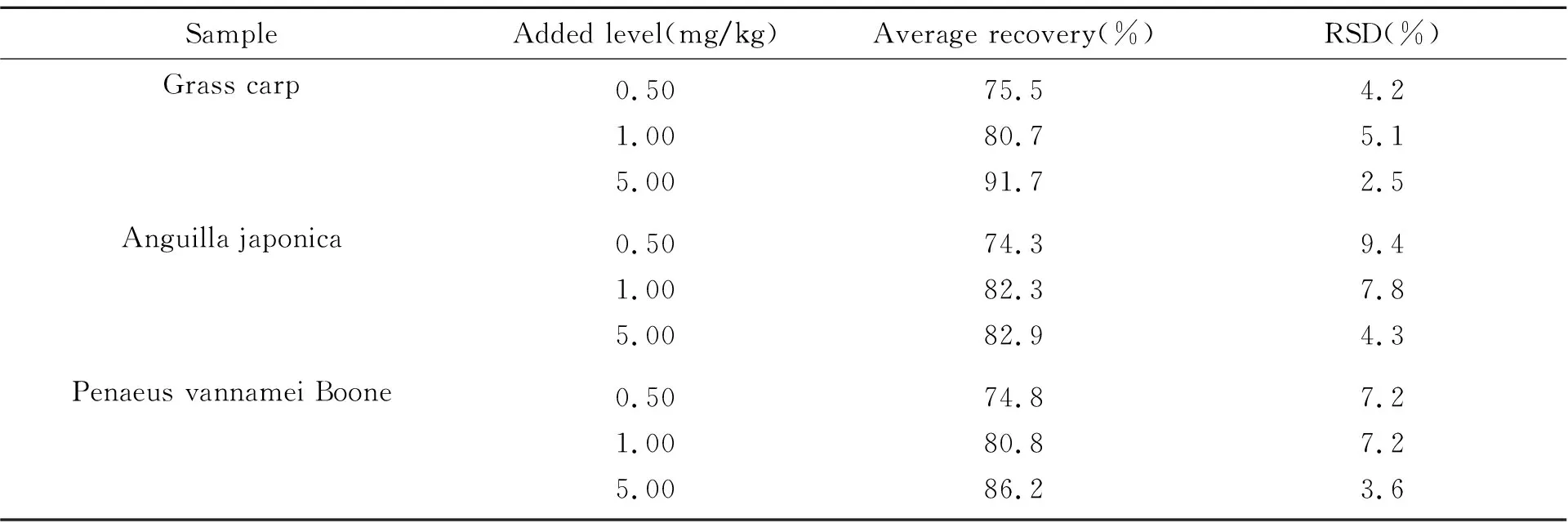
表1 冷冻水产品中乙萘酚的添加回收率及精密度
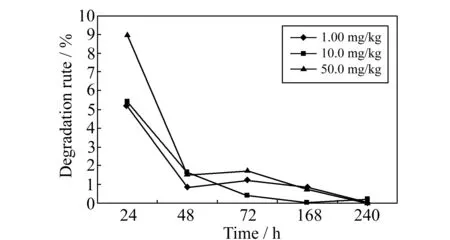
图1 乙萘酚的降解率曲线图Fig.1 Degradation rate of 2-naphthal
2.7 乙萘酚降解试验
在空白样品中添加1.00、10.0、50.0 mg/kg 3个浓度水平乙萘酚,每个浓度做5个平行,进行冷冻水产品中乙萘酚的降解试验。对样品进行24、48、72、168 h和240 h取样测定,整个试验过程样品保持在闭光冷冻状态。实验表明1.00、10.0、50.0 mg/kg 3个浓度的降解速率按添加水平由高到低成比例关系,分别为50.0 mg/kg >10.0 mg/kg>1.00 mg/kg,降解率如图1所示,3个浓度在24 h均有明显下降,在48 h趋近平缓,之后物质的降解不明显,残留水平稳定。
3 结论
采用高效液相色谱-荧光检测法测定冷冻水产品中的乙萘酚残留,以酸化乙腈提取可有效地去除样品中蛋白质的影响,同时具有较高的提取效率;冷冻去脂和LC-NH2固相萃取组合净化处理的方式避免了液夜萃取存在的耗时长、成本高、有机试剂用量大等问题。方法简便、有效,能有效去除复溶液中脂肪类物质的干扰;采用Inertil ODS-SP C18柱作为固定相,以乙腈-水(50∶50,V/V)作为流动相,为本方法提供了高分离度。该方法简便、稳定、处理成本低、回收率高,能满足水产品中乙萘酚的残留检测。乙萘酚残留物稳定性试验采用空白样品添加,避光冷冻储存24、48、72、168和240 h后取样测定。发现在5 个时间段的测定结果,除在24 h时有一个明显下降之后,残留物的浓度变化不大,降解率不明显,表明乙萘酚残留物在冷冻避光的情况下稳定性好,具有持续的污染能力。
猜你喜欢
中华养生保健(2020年9期)2021-01-18 03:12:36
无机化学学报(2019年2期)2019-02-27 06:53:38
山东工业技术(2016年15期)2016-12-01 05:30:45
分析测试学报(2015年8期)2016-01-13 06:19:28
发明与创新(2015年21期)2015-02-27 10:39:11
应用化工(2014年7期)2014-08-09 09:20:27
水土保持通报(2014年5期)2014-06-09 08:27:10
郑州大学学报(工学版)(2014年6期)2014-03-01 04:21:28
无机化学学报(2014年8期)2014-02-28 17:32:33
杭州师范大学学报(自然科学版)(2013年1期)2013-10-28 05:04:19
