
双二茂铁基吡咯衍生物电荷交互通道
2015-08-15 08:33:40胡宇强韩利民内蒙古工业大学化工学院呼和浩特010051
物理化学学报 2015年2期
胡宇强 竺 宁 韩利民(内蒙古工业大学化工学院,呼和浩特010051)
双二茂铁基吡咯衍生物电荷交互通道
胡宇强竺宁韩利民*
(内蒙古工业大学化工学院,呼和浩特010051)
以二茂铁炔烃为原料通过“一锅法”环加成反应合成了一系列2,5-双二茂铁基-1-苯基-吡咯衍生物,包括:2,5-双二茂铁基-1-(3-三氟甲基苯基)-吡咯(1),2,5-双二茂铁基-1-(4-氟苯基)-吡咯(2),2,5-双二茂铁基-1-苯基-吡咯(3),2,5-双二茂铁基-1-(4-乙基苯基)-吡咯(4)和2,5-双二茂铁基-1-(4-乙氧基苯基)-吡咯(5),使用元素分析,傅里叶变换红外(FTIR)光谱,质谱(MS)和核磁共振(NMR)等手段对化合物进行了结构表征.采用循环伏安法(CV),密度泛函理论(DFT)模拟计算研究了苯基上取代基对双二茂铁间电荷交互的影响.研究发现第一氧化电位(Ea1),峰电位差(ΔE)与取代基的哈米特常数(σ),吡咯1H NMR的化学位移(δ),吡咯N原子自然轨道(NBO)电荷之间存在显著线性关联;同时发现,N原子电荷密度升高,双二茂铁间电荷交互能力减弱,N原子电荷密度降低,双二茂铁间电荷交互能力提高.因此这类双二茂铁基吡咯衍生物中N原子电荷密度对双二茂铁间电荷交互起着关键的影响作用.
双二茂铁基;吡咯衍生物;电荷交互通道
www.whxb.pku.edu.cn
The project was supported by the Natural Science Foundation of Inner Mongolia,China(2012ZD01),Natural Science Foundation of Inner Mongolia University of Technology,China(X201207),and Graduate Student Research Innovation Foundation of Inner Mongolia,China(B20131012802).
内蒙古自治区自然科学基金(2012ZD01),内蒙古工业大学自然科学基金(X201207)和内蒙古自治区研究生科研创新基金(B20131012802)资助项目©Editorial office ofActa Physico-Chimica Sinica
1 引言
二茂铁具有独特的夹心配位结构,正二价铁离子处在两个平行的环戊二烯阴离子之间,当二茂铁失去一个非键轨道电子后,其空间结构不发生变化,从而表现出非常优异的电化学氧化还原稳定性.1,2凭借着这样的氧化还原特性,二茂铁衍生物已成为近几十年来金属有机化学和电化学领域的研究热点.3-8利用二茂铁优良的电化学特性,研究和制备二茂铁基分子电子器件(分子导线,分子马达、分子电子传感器等)尤为引人关注,9-11那些通过炔桥,烯桥连接的线型多二茂铁基共轭衍生物(如Fc―C≡C―Fc、Fc―CH=CH―Fc、Fc―C≡C―C≡C―Fc、Fc―(C≡C)n―Fc等12-15)已在分子导线研究领域得到应用.此外,π共轭多二茂铁基衍生物在分子催化、磁性、非线性光学材料等领域的应用前景也引起了人们的重视.其中二茂铁基羧酸过渡金属配合物16,17已经在催化领域得到应用.
研究分子电子器件的工作前提是了解分子中的电荷交互传递过程以及影响电荷交互的因素.18-21所以近年来,学术界开始重视对非线型共轭多二茂铁基衍生物的研究,22-25原因在于非线型共轭结构中各二茂铁基(Fc)之间的相互作用存在不同的偶合通道或者说整个体系存在几种类型的电荷交互传递路径,且不同的电荷传递通道之间存在着明显的相互作用和影响.本课题组26-32在之前的工作中研究了线性多二茂铁炔烃、烷烃衍生物以及非线性的多二茂铁基共轭六元环的电化学性质,发现二茂铁间的电子交互作用与连接键性质、取代基类型、分子空间构型、中心分子的平面性、杂原子桥等因素高度相关.与此同时,Tarraga33和Lang34-36等课题组也研究了双二茂铁取代五元杂环电荷交互作用,同样发现影响电荷交互作用的因素包含取代基性质、杂原子桥与碳原子桥的性质.
本文采用环加成反应31设计并合成了一系列二茂铁基取代的吡咯衍生物(1-5),包括2,5-双二茂铁基-1-(3-三氟甲基苯基)-吡咯(1)、2,5-双二茂铁基-1-(4-氟苯基)-吡咯(2)、2,5-双二茂铁基-1-苯基-吡咯(3)、2,5-双二茂铁基-1-(4-乙基苯基)-吡咯(4)和2,5-双二茂铁基-1-(4-乙氧基苯基)-吡咯(5).这些模型化合物保持主连接结构不变的情况下,在与N原子相连苯环上引入不同的吸电诱导基团(―CF3,―F)或供电诱导基团(―CH2CH3,―OCH2CH3),通过循环伏安法(CV)对它们的电化学性质进行研究,了解取代基与电荷交互之间的关系.研究以吡咯中原子的电荷密度及CV数据为主线,用取代基的哈米特常数(σ)37量度电子效应,用原子的自然轨道(NBO)电荷38-41量度电荷密度,用CV数据来判断电荷交互作用.发现吡咯中N原子的电荷密度高时双二茂铁间电荷交互作用弱,N原子的电荷密度低时双二茂铁间电荷交互作用强,所以吡咯中的N原子是影响这类双二茂铁吡咯衍生物电荷交互作用的关键桥原子.
2 实验部分
2.1试剂与仪器
1,4-双二茂铁基-1,3-丁二炔依据文献42合成制备.3-三氟甲基苯胺、4-氟苯胺、苯胺、4-乙基苯胺、4-乙氧基苯胺、四正丁基六氟磷酸铵[NBu4][PF6],纯度均为分析纯,购自阿法埃莎(中国)化学有限公司.其他试剂包括:二氯甲烷、石油醚、氯化亚铜等,纯度均为分析纯,购自国药集团化学试剂有限公司.实验中所用溶剂使用前均经过严格的脱水脱氧处理.
使用美国Thermo Nicolet FTIR型红外光谱仪;德国Elementar var III型元素分析仪;瑞士Bruker avance III 500 FT-MHz型核磁共振仪;日本Shimadzu LCMS-2020型液质联用仪对化合物(1-5)进行结构表征.晶体结构测定选取0.2 mm×0.05 mm×0.05 mm(2)、0.4 mm×0.3 mm×0.01 mm(3)单晶,在德国Bruker SMART APEX CCD衍射仪上,用Mo-K(λ= 0.071073 nm)于296(2)K下收集化合物(2)衍射数据,用Cu-K(λ=0.154184 nm)于293(2)K下收集化合物(3)衍射数据,采用直接法和差值傅里叶技术解析结构,几何法确定氢原子位置,吸收校正采用SADABS程序,数据收集、处理和晶胞参数的确定及修正采用Bruker SAINT和Smart软件,结构计算和精修采用SHELXSL 97软件.使用上海辰华CHI600E型电化学工作站,采用循环伏安法(溶剂为CH2Cl2;分析物浓度为0.05 mol·L-1;支持电解质[NBu4][PF6]浓度为0.1 mol·L-1;温度为25°C;扫描速率为100 mV·s-1;工作电极为Pt电极;参比电极为Ag/Ag+电极;对电极为Pt丝)对化合物(1-5)的电化学性质进行研究.使用B3LYP函数和6-311++G**基组对化合物(1-5)的结构进行优化和密度泛函理论(DFT)量子化学计算.
2.2双二茂铁基吡咯衍生物的合成方法
将1,4-双二茂铁基-1,3-丁二炔(210.2 mg,0.5 mmol),取代苯胺(2.5 mL,27.4 mmol)及Cu2Cl2(2.4mg,0.024 mmol)加入到密封反应器中,反应物在纯Ar气环境下100°C搅拌24 h,冷却至室温,减压抽除溶剂,得黑红色油状物.硅胶柱分离(200-300目中性硅胶,Φ2.0 cm×15 cm),二氯甲烷/石油醚(体积比为1:4)淋洗,第一带为未反应的1,4-双二茂铁基-1,3-丁二炔;第二带为产物.化合物(2,3)晶体采用二氯甲烷/石油醚(体积比为1/10)混合溶剂在低温下(-15°C)培养获得.
2,5-双二茂铁基-1-(3-三氟甲基苯基)-吡咯(1):得190 mg黄色固体,产率:65.7%.熔点(m.p.):235-237°C;1H NMR(CDCl3,500 MHz)δ:7.25-7.71(m,4H),6.42(s,2H),3.83-4.04(m,18H);13C NMR(CDCl3,500 MHz)δ:140.42,133.07,132.16,129.19,126.73,122.49(C6H4),125.00(CF3),108.79(C4H2N),78.68 (Ci-C4H2N)(Ci为与环戊二烯(Cp)连接的C原子),77.27,77.01,76.76,69.40,67.64,67.26(Cp);IR (KBr)ν:3093,3069,1594,1497,1454,1349,1318,1182,1127,1065,1003,808,770 cm-1;MS(ESI)m/ z:理论值为579.0,测得值为579.1(M+,100%);元素分析(%)C31H24F3NFe2:理论值为C,64.28;H,4.18;N,2.42.测得值为C,64.46;H,4.44;N,2.35.
2,5-双二茂铁基-1-(4-氟苯基)-吡咯(2):得180 mg黄色固体,产率:68.0%.熔点(m.p.):240-242°C;1H NMR(CDCl3,500 MHz)δ:7.14-7.26(m,4H),6.39 (s,2H),3.87-4.03(m,18H);13C NMR(CDCl3,500 MHz)δ:161.37,135.94,132.26,131.37,115.79,115.61(C6H4),108.20(C4H2N),78.91(Ci-C4H2N),77.27,77.01,76.76,69.41,67.53,66.91(Cp);IR(KBr)ν:3093,1602,1513,1415,1104,1003,835,816 cm-1;MS(ESI)m/z:理论值为529.0,测得值为529.1(M+,100%);元素分析(%)C30H24FNFe2:理论值为C,68.09;H,4.57;N,2.65.测得值为C,67.78;H,4.94;N,2.53.
2,5-双二茂铁基-1-苯基-吡咯(3):得165 mg黄色固体,产率:64.7%.熔点(m.p.):192-195°C;1H NMR(CDCl3,500 MHz)δ:7.31-7.50(m,5H),6.38 (s,2H),3.84-4.01(m,18H);13C NMR(CDCl3,500 MHz)δ:140.03,129.91,128.74,128.64(C6H5),107.96 (C4H2N),79.08(Ci-C4H2N),77.26,77.01,76.75,69.39,67.43,66.64(Cp);IR(KBr)ν:3089,3050,1602,1497,1419,1104,1000,820,769,695 cm-1;MS(ESI)m/z:理论值为511.0,测得值为511.3(M+,100%);元素分析(%)C30H25NFe2:理论值为C,70.48;H,4.93;N,2.74.测得值为C,70.49;H,5.44;N,2.66.
2,5-双二茂铁基-1-(4-乙基苯基)-吡咯(4):得165 mg黄色固体,产率:61.2%.熔点(m.p.):236-240°C;1H NMR(CDCl3,500 MHz)δ:7.22-7.32(m,4H),6.36(s,2H),3.87-4.01(m,18H),2.77-2.82(m,2H),1.33-1.36(m,3H);13C NMR(CDCl3,500 MHz)δ:144.91,137.52,132.28,129.63,128.09(C6H4),107.80(C4H2N),79.19(Ci-C4H2N),77.27,77.01,76.76,69.39,67.40,66.52(Cp),28.59(CH2),15.44(CH3);IR (KBr)ν:3089,2964,2921,1513,1419,1104,1003,843,761 cm-1;MS(ESI)m/z:理论值为539.1,测得值为539.2(M+,100%);元素分析(%)C32H29NFe2:理论值为C,71.27;H,5.42;N,2.60.测得值为C,70.81;H,5.18;N,2.53.
2,5-双二茂铁基-1-(4-乙氧基苯基)-吡咯(5):得181 mg黄色固体,产率:56.1%.熔点(m.p.):217-219°C;1H NMR(CDCl3,500 MHz)δ:6.99-7.25(m,4H),6.34(s,2H),4.13-4.15(m,2H),3.89-4.03(m,18H),1.49-1.52(m,3H);13C NMR(CDCl3,500 MHz)δ:159.09,132.65,130.82,114.36(C6H4),107.63 (C4H2N),79.19(Ci-C4H2N),77.27,77.01,76.76,69.37,67.46,66.40(Cp),63.80(OCH2),14.86(CH3);IR (KBr)ν:3085,2984,2918,1610,1509,1104,1049,812,754 cm-1;MS(ESI)m/z:理论值:555.1,测得:555.2(M+,100%);元素分析(%)C32H29ONFe2:理论值为C,69.22;H,5.26;N,2.52.测得值为C,69.53;H,5.60;N,2.51.
3 结果与讨论
通过Cu2Cl2催化的“一锅法”环加成反应合成了一系列吡咯环桥联的双二茂铁化合物(图1),并对化合物(1-5)性质研究进行了详细讨论.
3.1化合物(1-5)合成与电化学测试
吡咯衍生物的合成方法主要包括:Negishi交叉偶联反应34-36和Paal-Knorr缩合反应,43,44这些反应底物适应性好、收率高,被广泛的应用于制备二茂铁基取代吡咯衍生物.但是,文中目标化合物(1-5)的设计初衷是通过改变吡咯中N位苯基上的取代基来调控双二茂铁间的电荷交换,如采用上述两类反应制备目标化合物,会导致合成路线复杂,总收率低等后果.所以采用以1,4-双二茂铁基-1,3-丁二炔和取代伯胺在Cu2Cl2的催化下一步环合生成吡咯45,46的环加成反应.
化合物(1-5)的电化学性质采用循环伏安法进行研究,实验表明每个化合物都在图谱中表现出两对独立的氧化还原峰,所有氧化还原峰电位都集中在0-0.8 V区间内,第一氧化电位(Ea1)集中在19-120 mV之间,第二氧化电位(Ea2)集中在344-390 mV之间(表1,图2),每一对氧化还原峰在不同扫描速率(50,80,100,150,200,250 mV·s-1)的阳极峰电流(Ipa)/阴极峰电流(Ipc)均约等于1,所以可以认定所有双二茂铁吡咯衍生物都发生了两次一步一电子47氧化还原反应.
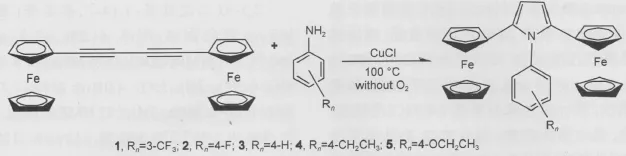
图1 化合物(1-5)的合成过程Fig.1 Synthesis processes of compounds(1-5)
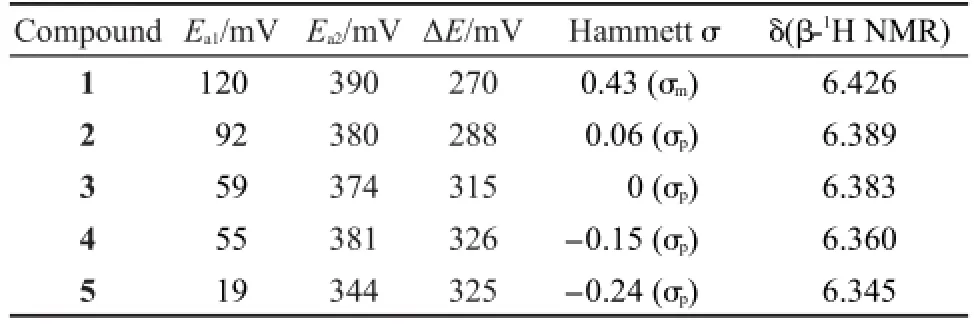
表1 化合物(1-5)表征数据及取代基Hammett常数(Hammett σ)Table 1 Characterize data and Hammett constant (Hammett σ)of compounds(1-5)
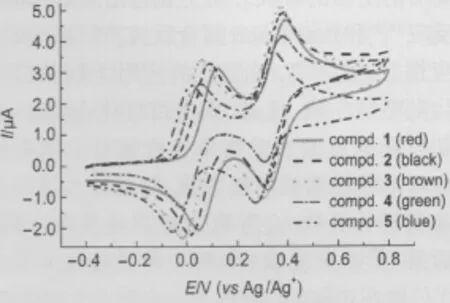
图2 化合物(1-5)循环伏安曲线Fig.2 Cyclic voltammograms of compounds(1-5)scan rate:100 mV·s-1
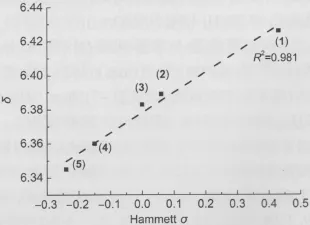
图3 化合物(1-5)吡咯环的1H NMR化学位移(δ)与取代基Hammett σ的相互关系Fig.3 Correlation of the1H NMR chemical shifts(δ)of the pyrrole ring and the Hammett σof compounds(1-5)
3.2吡咯环1H NMR化学位移
1H NMR谱图(参见Supporting Information)中可以找到化合物(1-5)所有的特征峰,其中吡咯环的1H NMR的化学位移(δ)分别是6.426(1)、6.389(2)、6.383(3)、6.360(4)和6.345(5)(表1).以吡咯环1H NMR位移(δ)为纵坐标,取代基Hammett常数(Hammett σ)为横坐标作图(图3).Hammett σ作为苯环上取代基电子效应标尺用于衡量取代基吸供电诱导效应,取代基从吸电基团变化到供电基团时,σ从正值呈线性变化到负值.37芳香性(共轭效应)被定义为维持抗磁环电流的能力,判断芳香性大小的最直接证据就是δ,δ取决于该质子周围的电子云密度,围绕或部分围绕质子的电子云密度越大,化学位移越向高场移动(δ更小),体系芳香性(共轭效应)越大.48化合物(1-5)取代基性质由供电性向吸电性变化时,Hammett σ由负值向正值变化,取代基的电子效应通过连接结构传递至吡咯环,使吡咯质子周围的电子云密度减小,δ变大,从图3可以发现这两个变量之间存在一次函数关系,确定系数R2=0.981,说明它们之间存在明显的线性关系.49,50化合物(1-5)的δ依次向高场移动,说明吡咯环共轭效应依次增大,电荷密度也随之增大,这一变化归因于取代基Hammett σ的变化,取代基的供电效应或吸电效应通过N原子传递至吡咯环.所以这时N原子起到了沟通吡咯环与苯环取代基电子效应的关键作用.
以第一氧化电位(Ea1)和峰电位差(ΔE)为纵坐标,吡咯的δ为横坐标作图(图4).δ逐渐增大(向低场移动,共轭性降低),理论上,π共轭作用增强,电荷交互作用增强;π共轭作用减弱,电荷交互作用减弱.51-53图4中δ变化趋势反映出π共轭作用减弱,Ea1增大,ΔE减小,呈现出了线性关系(Ea1,R2=0.913;ΔE,R2=0.865).但是,这一理论无法对Fc―CH=CH―Fc的电荷交互作用大于Fc―C≡C―Fc,即Fc―CH=CH―Fc(ΔE= 170 mV)>Fc―C≡C―Fc(ΔE=130 mV)54,55给出满意的解释.带着这样的疑问,我们深入研究了吡咯中四个C原子的NBO电荷.
3.3吡咯C原子对电荷交互的影响
采用量子化学计算方法获得化合物(1-5)中各原子的自然键轨道电荷.56-59计算使用Gaussian 03软件60中DFT混合泛函B3LYP算法获得化合物(1-5)的NBO电荷数值,56经计算得到NBO数值(图5,表2).
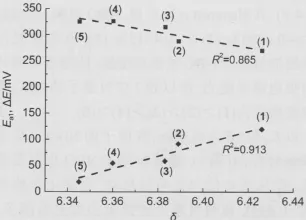
图4 化合物(1-5)吡咯1H NMR化学位移(δ)与Ea1(◆)和ΔE(■)值的相互关系Fig.4 Correlation of the1H-NMR chemical shifts(δ)of the pyrrole ring and the Ea1(◆)and ΔE(■)values of compounds(1-5)
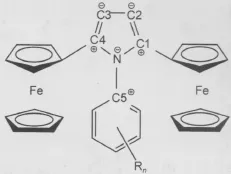
图5 化合物(1-5)吡咯环C原子编号Fig.5 C atomic numbers in pyrrole ring of compounds(1-5)
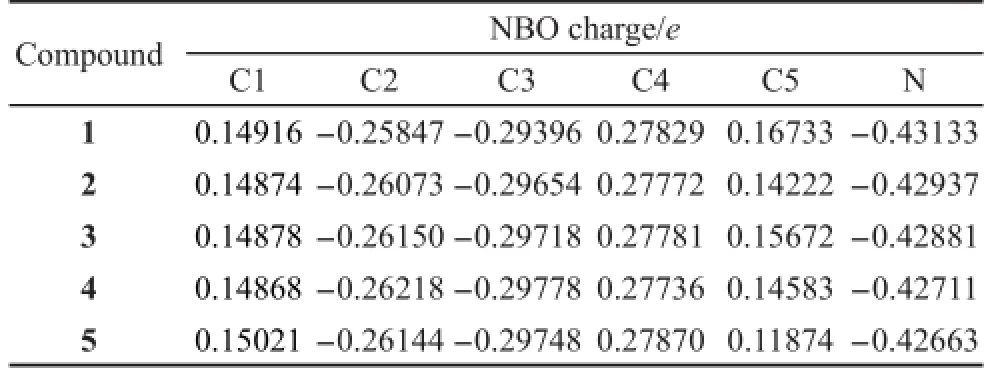
表2 化合物(1-5)NBO电荷数据Table 2 NBO charges of compounds(1-5)
化合物(1-5)吡咯环中的C原子NBO电荷密度分布呈现出两组不同趋势,与二茂铁相连的C原子(C1,C4)的NBO电荷为正值,未被取代的C原子(C3,C4)的NBO电荷为负值.理论上,如果化合物中原子X与供电基团相连时,受到供电诱导效应影响,NBO电荷为正值;当与吸电基团相连时,受到吸电诱导效应影响,NBO电荷为负值.57从表2数据分析,二茂铁基作为一个供电基团其供电效应使C1和C4的NBO电荷呈现正电性,而未被取代的C2与C3 的NBO电荷呈现负电性.以C原子的NBO电荷为纵坐标,取代基Hammett σ为横坐标分别对C1、C2、 C3、C4的NBO电荷值作图(图6),将每一个C原子的NBO电荷与取代基Hammett σ做线性拟合,确定系数R2都小于0.2,他们之间一次函数关系不明显,所以不存在线性关系,由此证明取代基的吸供电效应对吡咯中C原子的电荷密度没有直接的影响.
以Ea1和ΔE为纵坐标,C1、C2、C3、C4原子NBO电荷为横坐标分别作图(图7),C原子的NBO电荷与Ea1和ΔE对应坐标点散乱分布,不存在线性关系.证明吡咯中C原子电荷密度对二茂铁间电荷交互作用不存在直接影响.
从晶体结构(Supporting Information)角度来分析,化合物(2,3)的二茂铁基与吡咯环处于不同平面,化合物(2)中环戊二烯(C11―C12―C13―C14―C15)与吡咯环(N1―C7―C8―C9―C10)所成二面角为47.93°;环戊二烯(C21―C22―C23―C24―C25)与吡咯环(N1―C7―C8―C9―C10)所成二面角为45.49°;化合物(3)中环戊二烯(C15―C16―C17―C18―C19)与吡咯环(N1―C11―C12―C13―C14)所成二面角为40.11°;环戊二烯(C1―C2―C3―C4―C5)与吡咯环(N1―C11―C12―C13―C14)所成二面角为47.35°,说明二茂铁基与吡咯环的π共轭结构没有很好地发生叠加,所以二茂铁基体现出供电基团的供电诱导效应,以一个整体给吡咯环提供电荷.吡咯的δ与Ea1和ΔE的线性相关仅反映出吡咯环整体电荷密度与电荷交互之间的联系.
3.4吡咯环中N原子对电荷交互的影响
采用Gaussian 03软件中DFT混合泛函B3LYP算法获得了化合物(1-5)吡咯环中N原子的NBO电荷分布(图5,表2),N原子的NBO电荷分别为:-0.43133e(1)<-0.42937e(2)<-0.42881e(3)<-0.42711e(4)<-0.42663e(5).以N原子NBO电荷为纵坐标,取代基Hammett σ为横坐标作图(图8),发现随着取代基从供电性向吸电性转变,Hammettσ由负值向正值变化,其电子效应对N原子NBO电荷影响体现为负电荷密度逐渐增大,呈显著地线性关系(R2=0.966).

图6 化合物(1-5)吡咯环C原子NBO电荷与取代基Hammett σ的相互关系Fig.6 Correlation of the NBO charges of the C atom in pyrrole and the Hammett σof compounds(1-5)
从晶体结构(Supporting Information)角度来分析,化合物(2,3)的苯基与吡咯环处于不同平面,(2)中苯基(C1―C2―C3―C4―C5―C6)与吡咯环(N1―C7―C8―C9―C10)所成二面角为73.42°;(3)中苯基(C25―C26―C27―C28―C29―C30)与吡咯环(N1―C11―C12―C13―C14)所成二面角为66.97°.说明,苯基与吡咯环的π共轭结构没有发生叠加,所以化合物(1-5)中苯基体现诱导效应,而非共轭效应.
诱导效应理论,吸电诱导基团61使与其相连的原子带有部分的负电荷.C5的NBO电荷均为正值(表2),所以苯基整体成为一个吸电基团对吡咯发挥吸电诱导效应,改变苯基上取代基的吸供电效应可以调节苯环吸电诱导效应的强弱.以化合物(3)为基准,其取代基为H原子,Hammett σ=0,此时取代基为电中性;当取代基为供电诱导基团(4-OCH2CH3,4-CH2CH3),其Hammett σ为负值,NBO电荷-0.42663e (5)>-0.42711e(4)>-0.42881e(3),说明供电性取代基可以减少N原子NBO负电荷密度,即减弱苯环整体的吸电诱导能力;当取代基为吸电诱导基团(3-CF3,4-F),其Hammett σ为正值,NBO电荷-0.42881e (3)>-0.42937e(2)>-0.43133e(1),吸电性取代基可以增加N原子NBO负电荷密度,即增强了苯环整体的吸电诱导能力.所以表2中N原子的NBO负电荷密度顺序为(1)>(2)>(3)>(4)>(5).
以Ea1和ΔE为纵坐标,N原子的NBO电荷密度为横坐标作图(图9).随着N原子NBO负电荷密度降低,Ea1从高电位向低电位移动,呈现出正线性关系(R2=0.894),说明N原子所携带的负电荷越多,二茂铁越难失去非键轨道电子而发生氧化;当N原子携带的负电荷较少时,二茂铁基易于发生氧化.N原子NBO负电荷减少,ΔE增大,呈现出负线性关系(R2=0.902).说明N原子携带较多负电荷时(化合物1),两个二茂铁间的电荷交互作用减弱,ΔE只有270 mV,当N原子携带较少负电荷时(化合物4,5),ΔE增大至325-326 mV,二茂铁间的电荷交互作用得到增强.所以N原子所携带的负电荷大小就成为影响二茂铁基电荷交互作用强弱的关键因素,负电荷密度大,电荷交互作用弱;负电荷密度小,电荷交互作用强.
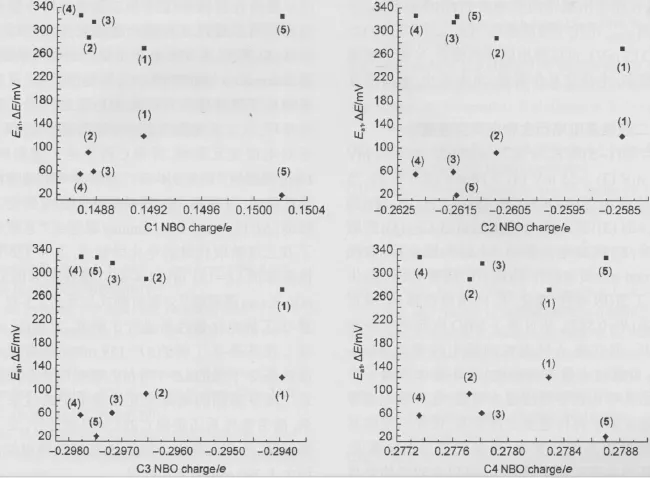
图7 化合物(1-5)Ea1(◆)和ΔE(■)值与吡咯环C原子NBO电荷的相互关系Fig.7 Correlation of the Ea1(◆)and ΔE(■)values and the C atomic NBO charges in pyrrole of compounds(1-5)
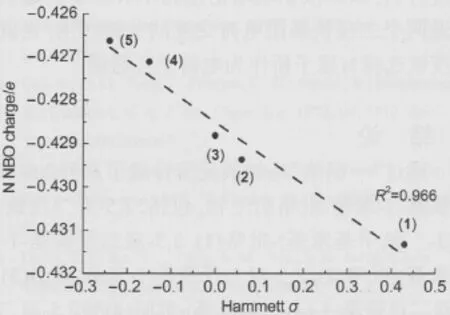
图8 化合物(1-5)N原子NBO电荷与取代基Hammett σ的相互关系Fig.8 Correlation of the N atomic NBO charges and the Hammett σof compounds(1-5)
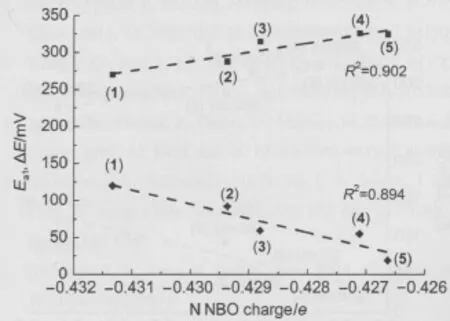
图9 化合物(1-5)Ea1(◆)和ΔE(■)值与吡咯环N原子NBO电荷的相互关系Fig.9 Correlation of the Ea1(◆)and ΔE(■)values of compounds(1-5)and the N atomic NBO charges in pyrrole
为了验证这一规律是否正确,我们幸运的发现Nemykin课题组62也完成了类似的二茂铁吡咯衍生物的合成与电化学研究工作,他们也合成了与化合物(3)结构相同的2,5-双二茂铁基-1-苯基-吡咯,ΔE也为315 mV,然而将苯基替换为甲基时,即2,5-双二茂铁基-1-甲基-吡咯,其ΔE增加到410 mV.以本文的观点,苯环在此体系中体现吸电诱导效应,而甲基(CH3,Hammett σ=-0.07)则体现供电诱导效应,所以N原子所携带的负电荷为NBO电荷(N-Ph)>NBO电荷(N-Me),电化学性质表明:ΔE(N-Me)(410 mV)>ΔE(N-Ph)(315 mV).可以得出同样的结论,N原子所携带负电荷多,电荷交互作用弱;负电荷少,电荷交互作用强.
3.5双二茂铁基吡咯衍生物电荷交互通道
化合物(1-5)的Ea1分别为:120 mV(1)>92 mV (2)>59 mV(3)>55 mV(4)>19 mV(5)(表1),当取代基从供电性(4-OCH2CH3(5),4-CH2CH3(4))到电中性(4-H(3))再到吸电性(4-F(2),3-CF3(1))的顺序变化时,Ea1向高电位移动.以Ea1为纵坐标,取代基Hammett σ为横坐标作图(图10).随着σ从负值(供电性)向正值(吸电性)变化,Ea1向高电位移动,成正线性关系(R2=0.885),从N原子NBO电荷密度分布数据分析,取代基从供电性向吸电性变化,Hammett σ从负值向正值方向移动,苯环整体作为一个吸电基团其吸电诱导效应逐步增强,这种吸电诱导效应通过N原子再传递至二茂铁基,促使二茂铁基的电荷密度由高到低变化.当取代基为供电基团,二茂铁基的电荷密度下降较少,可以在较低的外界电势下可以发生氧化,反之,氧化需要较高的外界电势.
化合物(1-5)的ΔE分别为270 mV(1),288 mV (2),315 mV(3),326 mV(4)和325 mV(5)(表1),以ΔE为纵坐标,取代基的Hammett σ为横坐标作图(图10),化合物(1-5)的ΔE与取代基的Hammett σ间存在负线性关系(R2=0.880).当σ从负值(供电性)向正值(吸电性)变化时,ΔE逐步减小,顺序为:325 mV (4-OCH2CH3(5))≈326 mV(4-CH2CH3(4))>315 mV (4-H(3))>288 mV(4-F(2))>270 mV(3-CF3(5)).
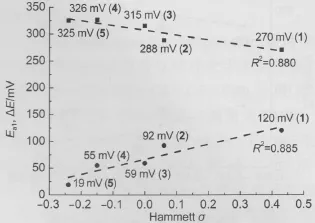
图10 化合物(1-5)的Ea1(●)和ΔE(■)值与取代基Hammett σ的相互关系Fig.10 Correlation of the Ea1(●)and ΔE(■)values and the Hammett σof compounds(1-5)
电荷在共轭非线性多二茂铁基化合物中的交互作用可以通过多种耦合通道来完成.63对于化合物(1-5),取代基从吸电基团变化到供电基团,取代基Hammett σ与吡咯的δ,Ea1呈线性关系.说明取代基的电子诱导效应可以有效地通过N原子传递到吡咯环,从宏观角度改变吡咯的电荷密度;研究C原子对电荷交互影响,发现C桥不是关键影响因素. Lang课题组36研究3,4-双二茂铁基-1H-吡咯的电化学性质时,发现两个二茂铁基团间的电荷交互作用较弱,ΔE仅为180 mV;Launay课题组64系统地研究了双二茂铁取代苯的电化学性质,其中1,2-双二茂铁基苯的ΔE=131 mV,1,4-双二茂铁基苯的ΔE=104 mV;Kwan课题组65分别对顺式与反式2,3-双二茂铁基-2-丁烯电化学性质进行了研究,发现(反式)2,3-双二茂铁基-2-丁烯的ΔE=159 mV,(顺式)2,3-双二茂铁基-2-丁烯的ΔE=170 mV.这些电化学数据说明双二茂铁基团的电荷交互作用不论通过杂芳香体系,碳芳香体系还是独立的C=C键进行,交互作用都很弱,不会出现化合物(1-5)这样的峰电位差(ΔE)均大于290 mV强交互作用.
研究N原子桥NBO电荷分布变化规律与电荷交互作用线性关系后,发现N原子中电荷密度对二茂铁基间的电荷交互能力有决定性影响.此外本课题组28发现,C桥原子的负电荷减少,也能提高二茂铁间电荷交互作用,当C桥原子的Mulliken电荷大小顺序为-0.1398e(Fc(CH3)C(C6H5)Fc)<-0.0978e (Fc(CH3)C(CH3)Fc)<-0.0473e(Fc(CH3)C(C2H5)Fc),ΔE(Fc(CH3)C(C6H5)Fc)(195 mV)<ΔE(Fc(CH3)C(CH3)Fc)(200 mV)<ΔE(Fc(CH3)C(C2H5)Fc)(220 mV).所以从原子的电荷分布与氧化还原电位之间相互关系角度分析,双二茂铁吡咯衍生物中N原子桥起到了沟通两个二茂铁基团电荷交互的关键作用,也就是二茂铁选择N原子桥作为电荷交互通道.
4 结论
通过“一锅法”环加成反应合成了系列2,5-双二茂铁基-1-苯基-吡咯衍生物,包括:2,5-双二茂铁基-1-(3-三氟甲基苯基)-吡咯(1),2,5-双二茂铁基-1-(4-氟苯基)-吡咯(2),2,5-双二茂铁基-1-苯基-吡咯(3),2,5-双二茂铁基-1-(4-乙基苯基)-吡咯(4)和2,5-双二茂铁基-1-(4-乙氧基苯基)-吡咯(5).以二茂铁作为电化学探针,利用循环伏安数据反映出取代基诱导效应对吡咯环电荷交互通道的影响,通过量子化学计算获得吡咯中各原子的电荷密度分布,综合晶体结构数据,发现N原子桥电荷密度对二茂铁电荷交互起到关键影响作用.桥原子电荷密度高,金属-金属间电荷交互作用弱;桥原子电荷密度低,金属-金属间电荷交互作用强.所以这类双二茂铁吡咯衍生物电荷交互选择N原子桥作为偶合通道.
Supporting lnformation: available free of charge via the internet at http://www.whxb.pku.edu.cn.
References
(1)Dunitz,J.D.;Orgel,L.E.;Rich,A.Acta Cryst.1956,9,373. doi:10.1107/S0365110X56001091
(2)Seiler,P.;Dunitz,J.D.Acta Cryst.1979,B35,1068.
(4)Gilbert,M.B.;Thomas,J.M.;Cowan,D.O.;Vanda,C.L.;Kaufman,F.;Roling,P.V.;Rausch,M.D.Inorg.Chem.1975,14,506.doi:10.1021/ic50145a011
(5)Pfaff,U.;Hildebrandt,A.;Schaarschmidt,D.;Ruffer,T.;Low,P.J.;Lang,H.Organometallics 2013,32,6106.doi:10.1021/ om4007533
(6)Dammer,S.J.;Solntsev,P.V.;Sabin,J.R.;Nemykin,V.N. Inorg.Chem.2013,52,9496.doi:10.1021/ic401163y
(7)Wang,J.;Feng,L.M.;Ma,F.J.;Lin,F.;Xie,L.L.;Yuan,Y.F. Chin.J.Org.Chem.2012,32,1479.[王静,冯丽敏,马飞娟,林芬,谢莉莉,袁耀锋.有机化学学报,2012,32,1479.]doi:10.6023/cjoc1112291
(8)Klimova,E.I.;Garcia,M.M.;Alamo,M.F.;Churakov,A.V.;Maya,S.C.;Beletskaya,I.P.Polyhedron 2014,68,272.doi:10.1016/j.poly.2013.11.003
(9)Morrison,J.W.H.;Krogsrud,S.;Hendrickson,D.N.Inorg. Chem.1973,12,1998.doi:10.1021/ic50127a009
(10)Richardson,D.E.;Taube,H.Coord.Chem.Rev.1984,60,107. doi:10.1016/0010-8545(84)85063-8
(12)Cowan,D.O.;Park,J.;Pittman,C.U.;Sasaki,Y.;Mukherjee,T. K.;Diamond,N.A.J.Am.Chem.Soc.1972,94,5110.doi:10.1021/ja00769a069
(13)Yamamoto,T.;Seki,K.;Yamamoto,A.;Motoyama,I.;Sano,H. Inorg.Chim.Acta 1983,73,75.doi:10.1016/S0020-1693(00)90829-6
(14)Dong,T.Y.;Ke,T.J.;Peng,S.M.;Yeh,S.K.Inorg.Chem. 1989,28,2103.doi:10.1021/ic00310a018
(15)Ribou,A.C.;Launay,J.P.;Sachtleben,M.L.;Li,H.;Spangler,C.W.Inorg.Chem.1996,35,3735.doi:10.1021/ic951376u
(16)Aguado,J.E.;Crespo,O.;Gimeno,M.C.;Jones,P.G.;Laguna,A.;Nieto,Y.Eur.J.Inorg.Chem.2008,3031.
(17)Carugo,O.;Santis,G.D.;Fabbrizzi,L.;Licchelli,M.;Monichino,A.;Pallavicini,P.Inorg.Chem.1992,31,765.doi:10.1021/ic00031a014
(18)Hollandsworth,C.B.;Hollis,J.W.G.;Slebodnick,C.;Deck,P. A.Organometallics 1999,18,3610.doi:10.1021/om990321n
(19)Camire,N.;Westerhoff,U.T.M.;Geiger,W.E.J.Organomet. Chem.2001,637,823.
(20)Tahara,K.;Akita,T.;Katao,S.;Kikuchi,J.I.Dalton Trans. 2014,43,1368.doi:10.1039/c3dt52503a
(21)Bruna,S.;Perles,J.;Nieto,D.;Ana,M.;Vadillo,G.;Cuadrado,I.J.Organomet.Chem.2014,751,769.doi:10.1016/j. jorganchem.2013.07.052
(22)Alvarez,J.;Kaifer,A.E.Organometallics 1999,18,5733.doi:10.1021/om990678r
(23)Qin,S.N.;Hu,J.R.;Chen,Z.L.;Liang,F.P.J.Coord.Chem. 2011,64,3718.doi:10.1080/00958972.2011.630072
(24)Javed,F.;Altaf,A.A.;Badshah,A.;Tahir,M.N.;Siddiq,M.;Rehman,Z.U.;Shah,A.;Ullah,S.;Lal,B.J.Coord.Chem. 2012,65,969.doi:10.1080/00958972.2012.664769
(25)Liu,Z.S.;Zhu,N.;Han,L.M.;Xie,R.J.;Hong,H.L.;Suo,Q. L.J.Coord.Chem.2012,65,2804.doi:10.1080/ 00958972.2012.704024
(26)Han,L.M.;Hu,Y.Q.;Suo,Q.L.;Luo,M.H.;Weng,L.H. J.Coord.Chem.2010,63,600.doi:10.1080/00958970903582688
(27)Xie,R.J.;Han,L.M.;Gao,Y.Y.;Zhu,N.;Hong,H.L.;Suo,Q. L.J.Coord.Chem.2012,65,4086.doi:10.1080/ 00958972.2012.732696
(28)Xie,R.J.;Han,L.M.;Suo,Q.L.;Hong,H.L.;Luo,M.H. J.Coord.Chem.2010,63,1700.doi:10.1080/ 00958972.2010.487102
(29)Xie,R.J.;Han,L.M.;Zhu,N.;Hong,H.L.;Suo,Q.L. J.Coord.Chem.2011,64,3180.doi:10.1080/ 00958972.2011.616198
(30)Xie,R.J.;Han,L.M.;Zhu,N.;Hong,H.L.;Suo,Q.L.;Fu,P. Polyhedron 2012,38,7.
(31)Hu,Y.Q.;Han,L.M.;Zhu,N.;Hong,H.L.;Xie,R.J.J.Coord. Chem.2013,66,3481.doi:10.1080/00958972.2013.841902
(32)Wang,Y.Q.;Han,L.M.;Suo,Q.L.;Zhu,N.;Hao,J.M.;Xie,R.J.Polyhedron 2013,54,221.doi:10.1016/j.poly.2013.02.043
(33)Tarraga,A.;Molina,P.;Curiel,D.;Velasco,M.D.Tetrahedron Letters 2002,43,8453.doi:10.1016/S0040-4039(02)02097-X
(34)Hildebrandt,A.;Schaarschmidt,D.;As,L.V.;Swarts,J.C.;Lang,H.Inorg.Chim.Acta 2011,374,112.doi:10.1016/j. ica.2011.02.058
(35)Hildebrandt,A.;Lang,H.Dalton Trans.2011,40,11831.doi:10.1039/c1dt10997a
(36)Hildebrandt,A.;Schaarschmidt,D.;Lang,H.Organometallics 2011,30,556.doi:10.1021/om100914m
(37)Hansch,C.;Leo,A.;Taft,R.W.Chem.Rev.199l,97,165.
(38)Nemukhina,A.V.;Weinhold,F.J.Chem.Phys.1993,98,1329.doi:10.1063/1.464299
(39)Badenhoopa,J.K.;Weinhold,F.J.Chem.Phys.1997,107,5406.doi:10.1063/1.474248
(40)Carpenter,J.E.;Weinhold,F.J.Chem.Phys.1988,92,4306.doi:10.1021/j100326a013
(41)Zimmerman,H.E.;Weinhold,F.J.Org.Chem.2013,78,1844.doi:10.1021/jo301620k
(42)Drake,S.R.;Khattar,R.Organomet.Synth.1988,4,234.
(43)Han,L.M.;Zhang,L.F.;Zhang,G.B.;Yin,Z.G.Chin.J. Inorg.Chem.2008,24,1807.[韩利民,张利峰,张广斌,尹志国.无机化学学报,2008,24,1807.]
(44)Sorensen,A.R.;Overgaard,L.;Johannsen,I.Synthetic Metals. 1993,55,1626.doi:10.1016/0379-6779(93)90296-9
(45)Imler,G.H.;Lu,Z.;Kistler,K.A.;Carroll,P.J.;Wayland,B. B.;Zdilla,M.J.Inorg.Chem.2012,51,10122.doi:10.1021/ ic300435t
(46)Feng,X.;Tong,B.;Shen,J.B.;Shi,J.B.;Han,T.Y.;Chen,L.;Zhi,J.G.;Lu,P.;Ma,Y.G.;Dong,Y.P.J.Phys.Chem.B 2010,114,16731.doi:10.1021/jp108254g
(47)Zheng,Q.W.;Hua,R.M.Tetrahedron Letters 2010,51,4512. doi:10.1016/j.tetlet.2010.06.092
(48)Smith,M.B.;March,J.MarchʹsAdvanced Organic Chemistry:Reactions,Mechanisms,and Structure,6th ed.;Wiley:New Jersey,2007;pp 54-55.
(49)Corry,A.J.;Goel,A.;Kenny,P.T.M.Inorg.Chim.Acta 2012,384,293.doi:10.1016/j.ica.2011.12.021
(50)Schaarschmidt,D.;Hildebrandt,A.;Bock,S.;Lang,H.Journal of Organometallic Chemistry 2014,751,742.doi:10.1016/j. jorganchem.2013.07.080
(51)Liu,S.G.;Perez,I.;Martin,N.;Echegoyen,L.J.Org.Chem. 2000,65,9092.doi:10.1021/jo001149w
(52)Schubert,C.;Wielopolski,M.;Mewes,L.H.;Rojas,G.D.M.;Pol,C.V.D.;Moss,K.C.;Bryce,M.R.;Moser,J.E.;Clark,T.; Guldi,D.M.Chem.Eur.J.2013,19,7575.doi:10.1002/ chem.201204055
(53)Justin,T.K.R.;Lin,J.T.;Wen,Y.S.Organometallics 2000,19,1008.doi:10.1021/om9908635
(54)Levanda,C.;Bechgaard,K.;Cowan,D.O.J.Org.Chem.1976,41,2700.doi:10.1021/jo00878a008
(55)Ribou,A.C.;Launay,J.P.;Sachtleben,M.L.;Li,H.;Spangler,C.W.Inorg.Chem.1996,35,3735.doi:10.1021/ic951376u
(56)Glendening,E.D.;Landis,C.R.;Weinhold,F.Journal of Computational Chemistry 2013,34,1429.doi:10.1002/ jcc.23266
(57)Weinhold,F.Journal of Computational Chemistry 2012,33,2363.doi:10.1002/jcc.v33.30
(58)Reed,A.E.;Weinhold,F.J.Chem.Phys.1985,84,1736.
(59)Reed,A.E.;Weinhold,F.J.Chem.Phys.1986,84,5687.doi:10.1063/1.449928
(60)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03,RevisionA.01;Gaussian Inc.:Pittsburgh,PA,2003.
(61)Smith,M.B.;March,J.MarchʹsAdvanced Organic Chemistry:Reactions,Mechanisms,and Structure,6th ed.;Wiley:New Jersey,2007;pp 19-20.
(62)Goetsch,W.R.;Solntsev,P.V.;Stappen,C.V.;Purchel,A.A.;Dudkin,S.V.;Nemykin,V.N.Organometallics 2014,33,145.doi:10.1021/om400901w
(63)Fink,H.;Long,N.J.;Martin,A.J.;Opromolla,G.;White,A.J. P.;Williams,D.J.;Zanello,P.Organometallics 1997,16,2646.doi:10.1021/om9701027
(64)Patoux,C.;Coudret,C.;Launay,J.P.;Joachim,C.;Gourdon,A. Inorg.Chem.1997,36,5037.doi:10.1021/ic970013m
(65)Chen,Y.J.;Pan,D.S.;Chiu,C.F.;Su,J.X.;Lin,S.J.;Kwan,K.S.Inorg.Chem.2000,39,953.doi:10.1021/ic991084j
Channel of Electronic lnteractions in Diferrocenyl Pyrrole Derivatives
HU Yu-QiangZHU NingHAN Li-Min*
(College of Chemical Engineering,Inner Mongolia University of Technology,Hohhot 010051,P.R.China)
2,5-Diferrocenyl-1-(3-trifluorom-ethylphenyl)-pyrrole(1),2,5-diferrocenyl-1-(4-fluorophenyl)-pyrrole (2),2,5-diferrocenyl-1-phenyl pyrrole(3),2,5-diferro-cenyl-1-(4-ethylphenyl)-pyrrole(4),and 2,5-diferrocenyl-1-(4-ethoxyphenyl)-pyrrole(5)were prepared by the one-pot cycloaddition reaction of ferrocenyl alkyne.The 2,5-diferrocenyl-1-phenyl-1-pyrrole derivatives were characterized by elemental analysis,Fourier-transform infrared(FTIR)spectroscopy,mass spectrometry(MS),and nuclear magnetic resonance(NMR)spectroscopy. The influence of substituents at the phenyl moiety on the electronic interaction was studied using cyclic voltammetry(CV)and density functional theory(DFT)calculations.Alinear relationship was observed between the first oxidation potential(Ea1),oxidation potential difference(ΔE)with Hammett constant(Hammett σ)of the substituent,pyrrole1H NMR chemical shift(δ),and pyrrole N natural bond orbital(NBO)charge.Ahigh N charge density weakened the electronic interaction,and vice versa.Electron transfer between the two ferrocenyl units of these diferrocenyl pyrrole derivatives was influenced by the N charge density.
Diferrocenyl;Pyrrole derivative;Channel of electronic interaction
August 5,2014;Revised:November 5,2014;Published on Web:November 6,2014.
O646
10.3866/PKU.WHXB201411061
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
农药科学与管理(2019年8期)2019-11-23 08:04:44
固体火箭技术(2016年5期)2016-11-03 05:35:29
合成化学(2015年10期)2016-01-17 08:56:06
中国塑料(2015年5期)2015-10-14 00:59:47
化学工业与工程(2015年1期)2015-02-10 03:01:33
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年9期)2014-02-28 17:33:11
无机化学学报(2014年7期)2014-02-28 17:32:28
