
取代基效应对N-4-取代苯亚甲基苯胺与N-4-取代苯亚甲基环己胺NMR和UV光谱影响的差异性
2015-08-15 08:33:37曹朝暾魏佰影曹晨忠湖南科技大学化学化工学院理论有机化学与功能分子教育部重点实验室分子构效关系湖南省普通高等学校重点实验室湖南湘潭411201
物理化学学报 2015年2期
曹朝暾 魏佰影 曹晨忠(湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,分子构效关系湖南省普通高等学校重点实验室,湖南湘潭411201)
[Article]
取代基效应对N-4-取代苯亚甲基苯胺与N-4-取代苯亚甲基环己胺NMR和UV光谱影响的差异性
曹朝暾魏佰影曹晨忠*
(湖南科技大学化学化工学院,理论有机化学与功能分子教育部重点实验室,分子构效关系湖南省普通高等学校重点实验室,湖南湘潭411201)
合成了N-4-取代苯亚甲基苯胺(1)与N-4-取代苯亚甲基环己胺(2)两个系列化合物,测定其13C和1H核磁共振(NMR)化学位移以及紫外(UV)吸收光谱.定量对比了取代基效应对两个系列化合物CH=N键的13C NMR化学位移δC(C=N)和1H NMR化学位移δH以及UV吸收光谱最大波长能量(νmax)的影响差异.研究结果表明,对于分子骨架相似的化合物(1)和(2),取代基效应的作用方式存在多样性:(i)化合物(1)的δC(C=N)、δH以及νmax受到基团的特殊交叉相互作用(Δσ2)的影响显著,而Δσ2对化合物(2)相应性能的影响很小;(ii)无论化合物(1)还是化合物(2),取代基场/诱导效应σF和共轭效应σR对δC(C=N)的影响为负相关,而对δH的影响为正相关,它们对δC(C=N)和δH的影响正好相反.另一方面,场/诱导效应σF对(1)和(2)的δC(C=N)影响重要,而对它们的δH影响很小;(iii)化合物(1)和(2)的δC(C=N)、δH以及νmax的变化规律,可分别建立通用方程表达,其中与CH=N的N原子键连苯基的影响可由指示变量(I)表示,该苯基对三种性能分别有固定的贡献.
苯亚甲基胺;取代基效应;核磁共振;紫外吸收;基团特殊交叉作用
www.whxb.pku.edu.cn
1引言
有机化合物的核磁共振(NMR)谱和紫外(UV)吸收光谱是化合物的重要物理特性,不仅用于有机化合物的分子结构解析,还常常用于分子结构-性能关系的理论研究.通常,取代基效应对分子内部电荷分布的影响,可采用NMR方法测定分子特定部位的C、H或其它原子的化学位移来估计.取代基对共轭有机分子前线轨道能量的影响,则可采用测定其UV吸收最大波长的方法确定.已知有机物的NMR和UV谱学性能与取代基效应密切相关.然而,取代基效应在化合物中所起的作用似乎表现出多样性,也比较复杂.“具有不同分子结构特征的有机化合物,取代基效应遵守什么规律?”一直是化学工作者试图探明的重要理论问题之一.比如,取代二苯乙烯XPhCH=CHPhY的UV吸收光谱最大波长(λmax)的能量(νmax)主要与取代基X、Y的激发态取代基参数 σCexC有关,1-6而取代N-苯亚甲基苯胺XPhCH=NPhY的UV吸收νmax则除了与取代基X、Y的激发态取代基参数σCexC有关之外,还与X、Y的基态极性参数(Hammette常数,σ)密切相关,同时还受基团X和Y特殊交叉作用的影响.7,8实际上这两类化合物是等电子体,只是链接两个苯环的桥键不同,一个是非极性的CH=CH键,另一个是极性的CH=N键.最近的研究表明,分子骨架的差别会导致取代基效应作用方式的改变.李富友等9研究了“推拉”型希夫碱染料的光化学和光电化学性能,得到很有意义的结果.Neuvonen等10曾用13C NMR方法对XPhCH=NPhY的C=N键中C原子电荷分布(qC或13C NMR化学位移δC(C=N))进行系统的研究,结果令人吃惊也令人感兴趣:(i)取代基X与Y对δC(C=N)影响正好相反,即吸电子的X基团与给电子的Y基团所起作用相同,给电子的X基团与吸电子的Y基团所起作用相同;(ii)存在取代基X与Y之间的特殊交叉作用.Cao等11-16进一步对有关化合物的δC(C=N)深入研究,提出以Δσ2定量表达取代基X与Y之间的特殊交叉作用.从以上分析得知,取代基在不同的分子骨架中作用方式不同,而在同一极性骨架分子XPhCH=NPhY的两端,取代基效应的作用则正好相反.这一现象使传统的取代基效应的理解受到挑战,它打破了吸电子取代基和给电子取代基的绝对概念,或许所谓吸电子取代基和给电子取代基只不过是特定分子骨架中观察到的特定现象.由此,我们想到一个更有趣的问题:如果分子骨架特征相同,只是共轭链长度有差异的情况下,取代基效应的作用方式会有差异吗?为探讨这一问题,我们选取分子结构非常相似的两个化合物系列,N-4-取代苯亚甲基苯胺(简写成XPhCH=NPh)和N-4-取代苯亚甲基环己胺XPhCH=N-c-C6H11(简写成XPhCH= NC6)为模型化合物,考察它们的取代基效应作用方式的异同.这两个化合物系列C=N键的N原子均与六元环相连,只不过前一系列的N原子与苯环连接可形成共轭,后一系列的N原子与环己基相连接不能发生共轭.
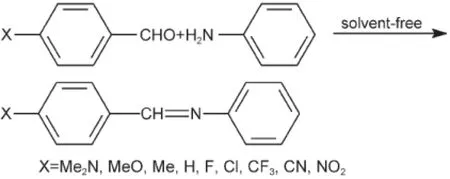
示意图1 化合物XPhCH=NPh的合成Scheme 1 Synthesis of compounds XPhCH=NPh
2 实验数据
N-4-取代苯亚甲基苯胺XPhCH=NPh(以下称化合物(1))的合成采用无溶剂方法,17见示意图1.粗产物经无水乙醇纯化,测定其1H NMR、13C NMR和UV吸收光谱.确认桥键CH=N上H原子的1H NMR化学位移δH和C原子的13C NMR化学位移δC(C=N),列于表1.10,18UV吸收光谱最大波长(λmax)的能量(νmax)列于表2.
N-4-取代苯亚甲基环己胺XPhCH=NC6(以下称化合物(2))的合成采用文献19提供的方法,见示意图2.粗产物经重结晶纯化,测定其1H NMR、13C NMR和UV吸收光谱.确认桥键CH=N上H原子的1H NMR化学位移δH和C原子的13C NMR化学位移δC(C=N),列于表1.UV吸收光谱最大波长(λmax)的能量(νmax)列于表2.

表1 化合物(1)和(2)桥键CH=N的13C NMR化学位移(δC(C=N))和1H NMR化学位移(δH)Table 1 13C NMR chemical shifts(δC(C=N))and1H NMR chemical shifts(δH)of CH=N bond in compounds(1)and(2)

表2 化合物(1)和(2)UV吸收光谱最大波长(λmax)及其能量(νmax)Table 2 UV absorption maximum wavelength(λmax)and its energy(νmax)of compounds(1)and(2)
3 结果与讨论
为了分析问题方便起见,我们把化合物(1)和(2)中涉及到的取代基X的取代基激发态常数(σex)5,20和CCHammett常数(σ)21列于表3.用这些参数分析取代基效应对化合物(1)和(2)的NMR和UV谱学性能影响的差异.
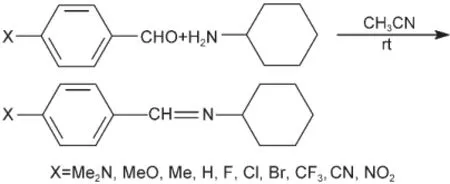
示意图2 化合物XPhCH=NC6的合成Scheme 2 Synthesis of compounds XPhCH=NC6
3.1取代基对NMR的影响差异
对于化合物(1)和(2),最容易确认的NMR化学位移是桥键CH=N的13C化学位移δC(C=N)和1H化学位移δH,它们对取代基效应的影响也比较敏感.因此,我们选定这两类原子的化学位移来比较取代基效应作用方式的差异.
3.1.1取代基对13C NMR的影响差异
根据我们以往的研究,16首先采用方程(1)作为模型,对化合物(1)和(2)的δC(C=N)(见表1)进行相关分析,得到方程列于表4.
δC(C=N)=a+bσF+cσR(1)
从表4中方程(1a)和(1b)可以看出,就δC(C=N)与取代基电子效应参数σF(场/诱导效应)和σR(共轭效应)的相关性而言,化合物(1)的相关性要比化合物(2)的相关性差.可能原因是化合物(1)桥键CH=N的N原子链接苯基,使得桥键两端的基团相互共轭,结果N原子上的苯基与X基团存在基团的特殊交叉作用(Δσ2).10,16因而,我们试图在方程(1)添加一项Δσ2,即用方程(2)进行回归,观察其结果是否有改进.方程(2)中Δσ2的取值方式为:对于化合物(1),N原子与苯基相连Δσ2=(σp(X)-(-0.01))2;化合物(2),N原子与环己基相连,Δσ2=(σp(X)-(-0.15))2.回归结果见表4的方程(2a)和(2b).
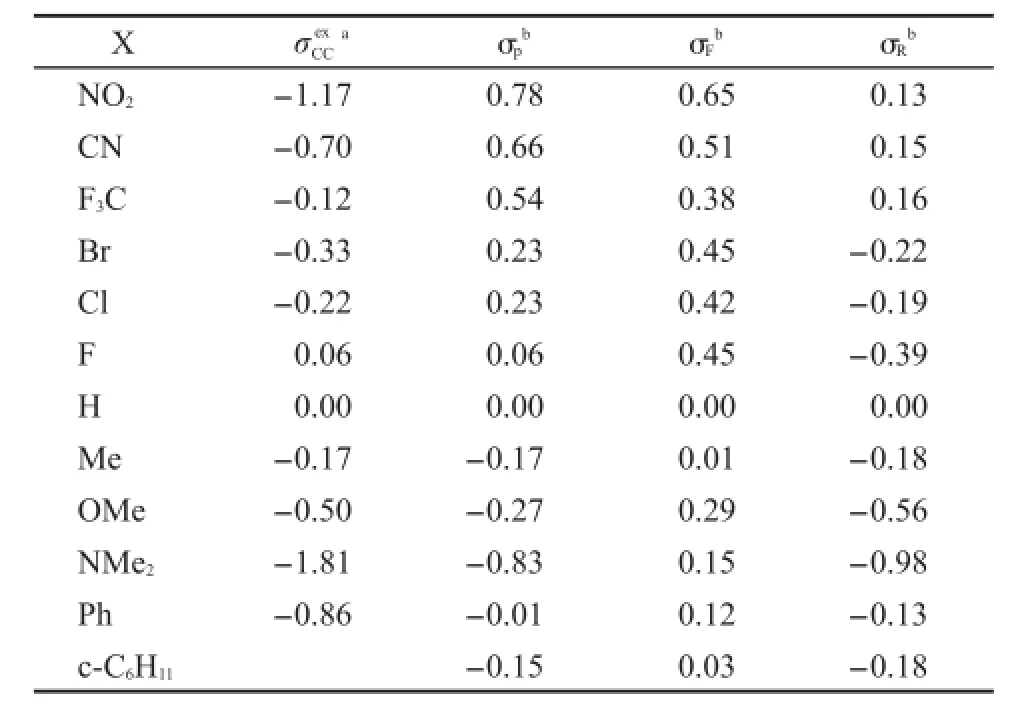
表3 一些取代基X的取代基效应常数Table 3 Substituent effect constants for some groups X
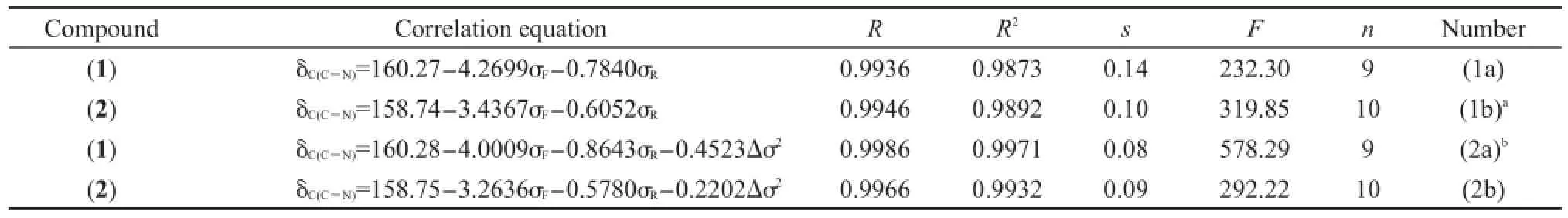
表4 化合物(1)和(2)的δC(C=N)相关分析结果Table 4 Results of correlation analysis of δC(C=N)for compounds(1)and(2)
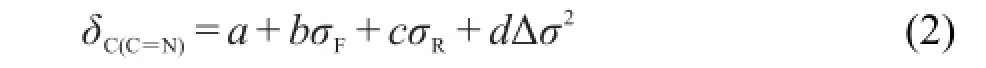
将表4的方程(2a)与(1a)进行比较,从标准偏差s和F值综合考虑,方程(2a)比(1a)有很大改进,说明Δσ2对化合物(1)的δC(C=N)有重要影响.然而,方程(2b)比(1a)没有实质改进,说明Δσ2对化合物(2)δC(C=N)的影响很小,可以忽略.我们注意到,表4中化合物(1)和(2)相对应的方程右边参数前面的系数具有相同的正负号,说明取代基X对两个系列化合物的δC(C=N)影响趋势相同.
表4中化合物(1)δC(C=N)的定量方程截距比化合物(2)相应方程的截距大,可能是与CH=N键N原子链接的苯基C原子(sp2杂化)比环己基C原子(sp3杂化)电负性大,同时苯基又可与N原子共轭的缘故.由此,我们设想是否可以采用一个指示变量(I)来表达苯基与环己基对δC(C=N)的影响差异,将化合物(1)和(2)两个系列的δC(C=N)变化规律用一个通式,即方程(3)表达.
δC(C=N)=a+bσF+cσR+dΔσ2+eI(3)
应该注意,根据前面的分析,Δσ2对化合物(2)的δC(C=N)贡献可以忽略,故采用方程(3)回归时其Δσ2取值为0.将表1中化合物(1)和(2)所有δC(C=N)值代入方程(3)回归,得到方程(4).
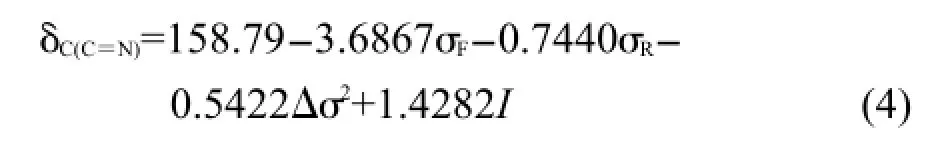
R=0.9953,R2=0.9906,s=0.13,F=367.25,n=19
方程(4)依然有很好的相关性,标准偏差为0.13,在实验测定误差范围内,说明用指示变量表达苯基与环己基对δC(C=N)的影响差异是可行的,该差异大约1.43.图1是方程(4)计算值δC(C=N),cal.对测定值δC(C=N),exp.作图.

图1 化合物(1)和(2)的计算值δC(C=N),cal.对测定值δC(C=N),exp.的相关图Fig.1 Plot of calculated δC(C=N),cal.versus experimental δC(C=N),exp.of compounds(1)and(2)
3.1.2取代基对1H NMR的影响差异
从分子骨架观察,化合物(1)和(2)的取代基X距离CH=N键比较远,取代基的立体效应对该化学键的13C和1H NMR化学位移的影响较小.根据取代基电子效应理论,人们通常会经验地认为如果CH=N键上C原子13C NMR化学位移δC(C=N)大,其H原子1H NMR的化学位移δH也大.事实果真如此吗?我们将化合物(1)和(2)的δH对相应的δC(C=N)作图,得到图2.令人吃惊的是,δH和δC(C=N)两者之间并不存在线性关系.按照前面3.1.1节的方法,我们首先分析δH与取代基电子效应参数σF和σR的相关,结果列于表5.从表5方程(5)和(6)看出,化合物(1)和(2)两者的δH均与σF和σR有良好的相关性,而且是正相关(参数前面的系数为正);然而,化合物(1)和(2)两者的δC(C=N)与σF和σR呈负相关(见表4方程(1a)和(1b)),这说明取代基X的电子效应(σF和σR)对CH=N键C原子的δC(C=N)和H原子的δH影响正好相反,确实超出了人们的经验预期,也是图2没有线性关系的真正原因.从方程(5)和(6)中参数σF和σR前面的系数比值(绝对值),可以认为场/诱导效应σF的贡献较小.若忽略该项重新回归,得到方程(7)和(8)(见表5).从方程的标准偏差s和F值综合考虑,方程(7)和(8)与相应的方程(5)和(6)的结果没有显著的差异,说明场/诱导效应σF对δH的影响可以忽略.进一步添加Δσ2一项作双参数回归,得到表5的方程(9)和(10),其结果表明方程(9)比(7)有较大改进,而方程(10)比(8)没有实质改进.这也说明Δσ2仅对化合物(1)的δH有重要影响,对化合物(2)的δH影响很小.
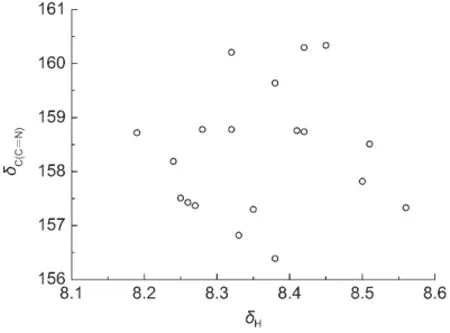
图2 化合物(1)和(2)的δC(C=N)对相应的δH的相关图Fig.2 Plot of δC(C=N)of compounds(1)and(2)versus their corresponding δH
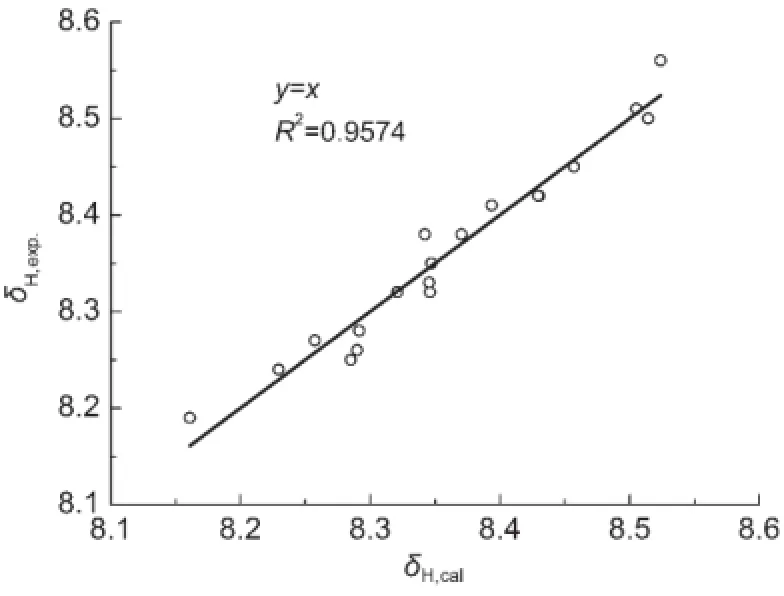
图3 化合物(1)和(2)的计算值δH,cal.对测定值δH,exp.的相关图Fig.3 Plot of calculated δH,cal.versus experimental δH,expof compounds(1)and(2)
比较表5的方程,化合物(1)的δH定量方程比相应化合物(2)的方程的截距稍大,可能是苯基和环己基分别与C=N的N相连的差异.若用指示变量I表示这一差异,同时将化合物(2)的Δσ2取值为0,采用式(11)作为通用模型方程对化合物(1)和(2)的δH值进行回归,得到方程(12).
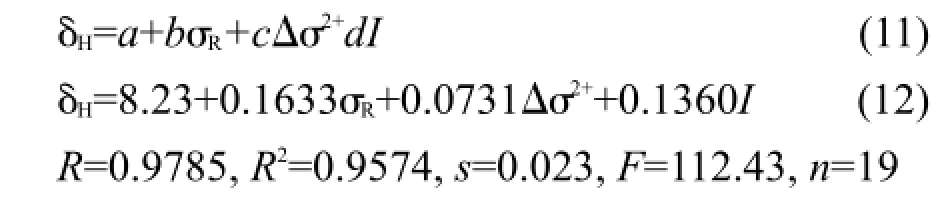
方程(12)的标准误差s只有0.023,在实验测定误差范围内,图3是δH的计算值对测定值的相关图.就方程的相关系数而言,似乎不是很好,其原因可能是这些化合物的δH值变化范围太小,最大值和最小值之间的间隔仅仅为0.37.
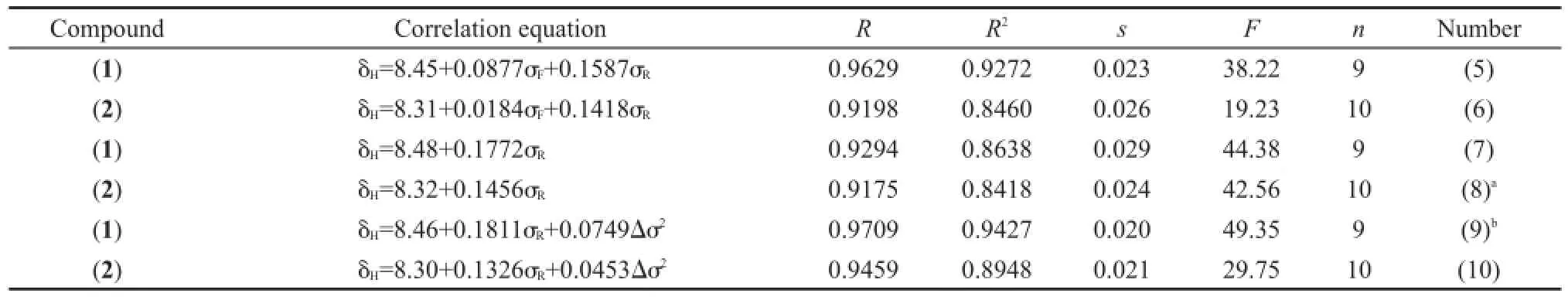
表5 化合物(1)和(2)的δH相关分析结果Table 5 Results of correlation analysis of δHfor compounds(1)and(2)
3.2取代基对UV吸收的影响差异
Cao等1,7最近提出激发态取代基常数σexCC,并用该参数对二苯乙烯类和共轭希夫碱类等化合物的UV吸收光谱进行定量相关,1-8得出二苯乙烯类化合物的UV吸收νmax主要受到取代基的σCexC参数影响,而共轭希夫碱类的UV吸收νmax则受到取代基σCexC参数和Hammett参数σp的共同影响.通过对化合物(1)和(2)UV吸收νmax影响因素的系统分析发现,化合物(1)的νmax可用方程(13)较好关联,而化合物(2)的νmax可用方程(14)较好关联.
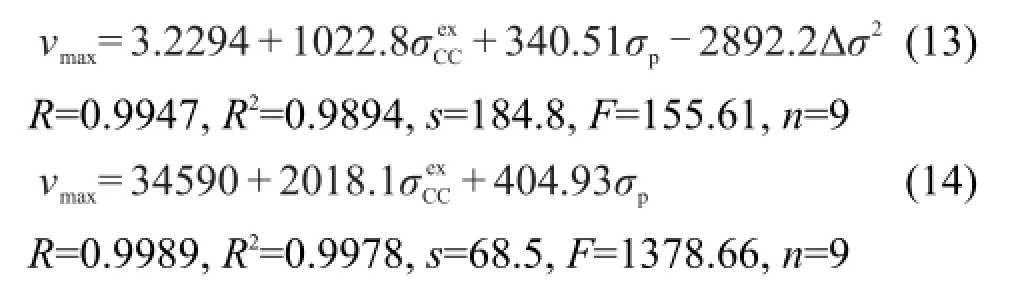
从方程(13)和(14)可以看出,基团的特殊交叉作用Δσ2对化合物(1)的νmax有重要影响,但对化合物(2)的νmax影响很小,可以忽略.
采用本文3.1节的方法,对化合物(1)添加指示变量I并将化合物(2)的Δσ2取值为0,就可以将化合物(1)和(2)的νmax建立统一的定量方程(15).该方程有很好的相关性,用方程(15)计算出νmax再换算成波长λmax,结果计算波长与测定波长的平均绝对偏差只有1.4 nm,最大绝对偏差只有2.9 nm.

表6 化合物(3)13C NMR的δC(C=N)值及回归结果Table 6 13C NMR δC(C=N)values and results of correlation analysis for compounds(3)
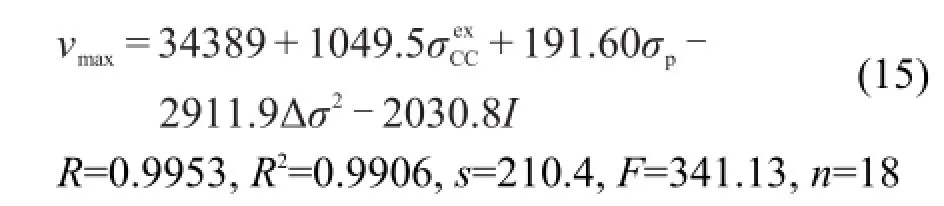
3.3分子聚集的可能影响
现代物理有机化学研究表明,有机化合物在溶液中受到溶剂极性、浓度等因素影响会发生聚集,从而影响化合物的NMR化学位移值、光物理性质及化学反应活性.22例如,二苯乙烯类化合物在环己烷、乙醚、三氯甲烷、乙腈和醇等10多种性能不同的溶剂中,紫外吸收最大波长能量就受到溶剂效应的显著影响,其中溶剂效应可用logP(溶剂在水/正辛醇中分配系数的对数)表达.23因而,为了避免溶剂效应的干扰,我们采用在同种溶剂中测定模型化合物的性能,即在氘代氯仿CDCl3中测定NMR化学位移值,在无水乙醇中测定UV吸收光谱.虽然溶剂效应基本消除,但是分子间的聚集作用不能消除,因为分子间的聚集在很低浓度下就可以发生.尤其是芳香烃,除了分子间的疏水亲酯作用外,还存在π-π堆积相互作用,24-26更容易发生分子间的聚集,并影响其NMR化学位移值及光物理性质.25,27,28比较化合物(1)和(2)的分子结构可以看出,化合物(1)在CH=N键两端都连接π电子共轭的苯环,而化合物(2)仅在CH=N键一端连接苯环,另一端连接环己基,环己基没有π电子体系,通常以椅式构象存在.因而化合物(1)比化合物(2)分子的π电子共轭体系更大,也更接近平面,更易于通过苯环之间的π-π作用发生聚集.很有可能,正是由于化合物(1)更易于聚集,使得化合物(1)在NMR化学位移值和UV吸收定量相关需要加入Δσ2一项,而化合物(2)的相应性能相关则可以忽略该项.
为了检验上述推断,我们选用与化合物(1)骨架相同,而取代基处于C=N键N原子连接的苯环上的化合物异构体PhCH=NPhX(以下称化合物(3))为模型.当基团X=NO2,CN,F,Cl,H,Me,OMe,NMe2时,它们的CH=N键δC(C=N)测定值分别为162.71、162.44、160.16、160.71、160.34、159.59、158.41和155.97.10对这些δC(C=N)值进行回归,得到表6的方程(16)和(17),结果表明加入Δσ2一项同样可获得更好的结果,见表6方程(17).
应该指出,由于本文的主要从有关化合物本身结构上的特点出发,考察取代基的电子效应对NMR化学位移值和UV吸收的影响,对分子聚集效应的影响并未作深入探讨.所涉及化合物分子聚集效应对NMR化学位移值和UV吸收的影响,是一个值得进行系统深入研究的课题,本文目前还不能对此得出具体的有价值的结论.
4 结论
通过上面对化合物(1)和(2)的NMR和UV吸收光谱的分析,可以得出如下结论:(i)尽管化合物(1)和(2)分子骨架结构相似,由于化合物(1)桥连键CH=N的N原子与苯基链接形成共轭,因而化合物(1)NMR谱的δC(C=N)、δH以及UV吸收谱的νmax受到基团特殊交叉作用Δσ2的影响显著;而Δσ2对化合物(2)相应性能的影响很小.(ii)无论化合物(1)还是化合物(2),取代基场/诱导效应σF和共轭效应σR对δC(C=N)的影响为负相关,而对δH的影响为正相关,两者正好相反.另一方面,场/诱导效应σF对(1)和(2)的δC(C=N)影响重要,而对(1)和(2)的δH影响却很小.这是对取代基效应作用所观察到的新结果,值得从理论上深入探索.(iii)可以将化合物(1)和(2)NMR谱的δC(C=N)、δH以及UV吸收谱的νmax分别建立通用方程,其中与CH=N的N原子键连苯基的影响可由指示变量I表示,该苯基对三种性能有固定的贡献.(iv)一方面,分子骨架结构类似的化合物,取代基效应的作用方式并不完全相同;另一方面,同一类分子骨架体系的不同谱学性能,取代基效应的作用也可能正好相反(比如对δC(C=N)和δH的影响).因而,取代基效应的作用是非常复杂的,其影响规律远远没有认识清楚,在研究类似化合物体系的具体性能时,对取代基效应不能盲目类推.included.This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
Supporting lnformation: The nuclear magnetic resonance (NMR)spectra of compounds XPhCH=N-c-C6H11have been
References
(1)Cao,C.;Chen,G.;Yin,Z.J.Phys.Org.Chem.2008,21(9),808.doi:10.1002/poc.v21:9
(2)Chen,G.;Cao,C.Chin.J.Chem.Phys.2009,22(4),366.doi:10.1088/1674-0068/22/04/366-370
(3)Chen,G.;Cao,C.J.Phys.Org.Chem.2010,23(8),776.
(4)Cao,C.;Chen,G.;Wu,Y.Sci.China Chem.2011,54(11),1735.doi:10.1007/s11426-011-4379-7
(5)Cao,C.;Sheng,B.;Chen,C.J.Phys.Org.Chem.2012,25(12),1315.doi:10.1002/poc.v25.12
(6)Cao,C.;Zhu,Y.;Chen,G.J.Phys.Org.Chem.2013,26(10),834.doi:10.1002/poc.v26.10
(7)Chen,G.;Cao,C.;Lu,B.;Sheng,B.J.Phys.Org.Chem.2012,25(4),327.doi:10.1002/poc.v25.4
(8)Cao,C.;Fang,Z.Spectrochim.Acta A 2013,111,62.doi:10.1016/j.saa.2013.03.082
(9)Li,F.Y.;Zheng,J.;Liu,T.T.;Jin,L.P.;Zhao,X.S.;Guo,J.Q. Acta Phys.-Chim.Sin.2000,16,787.[李富友,郑杰,柳汀汀,金林培,赵新生,郭建权.物理化学学报,2000,16,787.]doi:10.3866/PKU.WHXB20000906
(10)Neuvonen,H.;Neuvonen,K.;Fülop,F.J.Org.Chem.2006,71,3141.doi:10.1021/jo0600508
(11)Chen,G.;Cao,C.;Sheng,B.;Zhu,Y.;Wu,Z.;Wu,X.J.Phys. Org.Chem.2012,25(10),828.doi:10.1002/poc.v25.10
(12)Chen,G.;Cao,C.;Zhu,Y.;Wu,Z.;Wu,X.Spectrochim.Acta A 2012,99,218.doi:10.1016/j.saa.2012.09.028
(13)Fang,Z.;Cao,C.;Chen,G.J.Phys.Org.Chem.2012,25(12),1343.doi:10.1002/poc.v25.12
(15)Fang,Z.;Cao,C.;Wu,W.;Wang,L.J.Phys.Org.Chem.2013,26,249.doi:10.1002/poc.v26.3
(16)Cao,C.;Lu,B.;Chen,G.J.Phys.Org.Chem.2011,24(4),335.doi:10.1002/poc.1760
(17)Schmeyers,J.;Toda,F.;Boy,J.;Kaupp,G.J.Chem.Soc.Perkin Trans 2 1998,989.
(18)Lu,B.Substituent Effect on the Spectra and Molecular Configuration of4,4ʹ-DisubstitutedN-benzylidenebenzenamine Derivatives.Master Dissertation,Hunan University of Science and Technology,Hunan,2011.[卢冰涛.取代基效应对4,4ʹ-二取代氮苄叉苯胺衍生物光谱性能及分子构型的影响[D].湖南:湖南科技大学,2011.]
(19)Keglevich,G.;Kiss,N.Z.;Menyhárd,D.K.;Fehérvári,A.;Csontos,I.Heteroatom Chem.2012,23(2),171.doi:10.1002/ hc.v23.2
(21)Hansch,C.;Leo,A.;Taft,R.W.Chem.Rev.1991,91,165.doi:10.1021/cr00002a004
(22)Jiang,X.K.;Zhang,J.S.Aggregation and Self-Coling of Organic Molecules;Shanghai Science and Technology Press:Shanghai,1996;pp 94-112.[蒋锡夔,张劲松.有机分子的簇集和自卷.上海:上海科学技术出版社,1996:94-112.]
(23)Cao,C.Z.;Chen,G.F.;Wu,Y.X.Sci.Sin.Chim.2012,42(2),127.[曹晨忠,陈冠凡,武亚新.中国科学:化学,2012,42(2),127.]doi:10.1007/s11426-011-4379-7
(24)Shetty,A.S.;Zhang,J.;Moore,J.S.J.Am.Chem.Soc.1996,118,1019.doi:10.1021/ja9528893
(25)Jones,G.,II;Vullev,V.I.J.Phys.Chem.A 2001,105,6402. doi:10.1021/jp010087q
(26)Venkataramana,G.;Sankararaman,S.Org.Lett.2006,8(13),2739.doi:10.1021/ol060767h
(27)Goon,P.;Das,S.;Clemett,C.J.;Tiddy,G.J.T.;Kumar,V.V. Langmuir 1997,13,5577.doi:10.1021/la970502z
(28)Luca,G.D.;Romeo,A.;Scolaro,L.M.J.Phys.Chem.B 2005,109,7149.doi:10.1021/jp0448797
Effect of Substituents on the NMR and UV Spectra of N-(4-substituted benzylidene)Anilines and N-(4-substituted benzylidene)Cyclohexylamines
CAO Chao-TunWEI Bai-YingCAO Chen-Zhong*
(Hunan Provincial University Key Laboratory of QSAR/QSPR,Key Laboratory of Theoretical Organic Chemistry and Function Molecule of Ministry of Education,School of Chemistry and Chemical Engineering,Hunan University of Science and Technology,Xiangtan 411201,Hunan Province,P.R.China)
Two series of compounds:N-(4-substituted benzylidene)anilines(1)and N-(4-substituted benzylidene)cyclohexylamines(2)were synthesized.Their13C NMR and1H NMR chemical shifts and their UV absorption spectra were obtained.Compounds(1)and(2)were compared quantitatively to determine the effect of the substituents on the13C NMR chemical shifts δC(C=N)and the1H NMR chemical shifts δHof the CH=N bond,and the UV absorption maximum wavelength energies νmax.Our results show that the substituents affect compounds(1)and(2)differently despite them having a similar molecular skeleton.These effects are:(i)a substituent specific cross-interaction effect(Δσ2)that significantly affects the δC(C=N),δH,and νmaxof compounds (1)while its effect on the corresponding properties of compounds(2)is limited,(ii)for compounds(1)and compounds(2)the field/induced effect σFand the conjugation effect σRof the substituents negatively affect δC(C=N).However,theypositivelyinfluence δHandthusboth σFand σRshowoppositebehaviortoward δC(C=N)comparedwith δH.In contrast the field/induced effect greatly affects the δC(C=N)of both(1)and(2)but does not affect their δH,(iii)the regular change in δC(C=N),δH,and the νmaxof(1)as well as(2)can be expressed by a general equation in which the effect of the phenyl group attached to the N atom of the CH=N bond can be expressed by a dummy parameter I.The phenyl group has a constant contribution toward these three properties.
September 24,2014;Revised:December 19,2014;Published on Web:December 19,2014.
Benzylidene-aniline;Substituent effect;Nuclear magnetic resonance;Ultraviolet absorption;Substituent specific cross-interaction.
O641
10.3866/PKU.WHXB201412191
The project was supported by the National Natural Science Foundation of China(21272063,21072053)and Natural Science Foundation of Hunan Province,China(14JJ3112).
国家自然科学基金(21272063,21072053)和湖南省自然科学基金(14JJ3112)项目资助
©Editorial office ofActa Physico-Chimica Sinica
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 21:27:59
少儿科学周刊·儿童版(2021年22期)2021-12-11 06:42:32
农药科学与管理(2019年8期)2019-11-23 08:04:44
中国资源综合利用(2017年1期)2018-01-22 02:44:30
山东工业技术(2016年15期)2016-12-01 05:31:08
中国粮油学报(2016年5期)2016-01-23 02:44:53
化学工业与工程(2015年1期)2015-02-10 03:01:33
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:05
无机化学学报(2014年12期)2014-02-28 17:34:01
