
核壳结构Ni@Au/C用作DBFC阳极催化剂
2015-08-01 14:49梁建伟刘慧红卫慧凯卫国强段东红
电源技术 2015年10期
梁建伟,刘慧红,卫慧凯,卫国强,段东红
(太原理工大学洁净化工研究所,山西太原030024)
核壳结构Ni@Au/C用作DBFC阳极催化剂
梁建伟,刘慧红,卫慧凯,卫国强,段东红
(太原理工大学洁净化工研究所,山西太原030024)
采用连续两步化学还原法制备了Ni-Au摩尔比为1∶1的碳载核壳结构(Ni@Au/C)纳米粒子。通过X射线衍射(XRD)和透射电镜(TEM)对其结构进行表征,以循环伏安(CV)、计时电位(CP)、计时电流(CA)和电池测试技术对BH4-电化学氧化活性进行测试。结果表明,所制备的核壳结构纳米粒子直径在10 nm左右,其电化学活性面积为815.7 cm2/mg,约是实心纳米Au/C催化剂的5倍,且对BH4-电氧化反应显示出更好的催化活性。以Ni@Au/C为阳极催化剂,Pt网(1 cm×1 cm)为阴极组装的单电池在20℃时最大功率密度达到74 mW/cm2。
核壳结构纳米粒子;阳极催化剂;镍;金;电催化氧化;硼氢化物燃料电池
燃料电池是一种清洁、高效和安全的新型电池,以硼氢化物为燃料的硼氢化物燃料电池 (DBFC)具有理论能量密度高(9.3 Wh/g)、阳极反应动力学快和燃料易于储存和运输等优点,被认为是一种极有潜力的电池[1]。DBFC的阳极氧化反应如下:
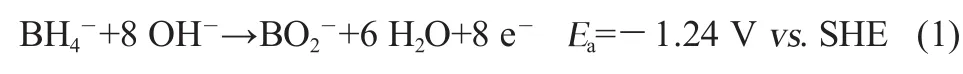
此反应为8 e-转移过程,但实际上由于BH4-的氧化过程十分复杂,只有很少的催化剂能使其实现8 e-转移。金(Au)作为一种高极化的贵金属,有较高的电子利用率,使BH4-的电氧化反应在较高电势区释放的电子数接近8,是一种高效的DBFC阳极电极材料[2-3]。但以单金属Au作为DBFC阳极催化剂时往往表现出较慢的动力学[4]。金属Ni具有较低的过电位和较好的极化性能,有利于提高DBFC的输出电压[5]。而且Ni的3 d轨道处于未充满状态,和Au形成核壳结构后,可能使壳层的Au原子具有某种特殊的性质,表现出更好的催化性能。
因此,本文利用反相微乳液体系的均相分散作用,采用连续两步化学还原法制备了Ni-Au摩尔比为1∶1的碳载核壳结构(Ni@Au/C)纳米粒子。利用透射电镜(TEM),X射线衍射光谱(XRD)等对制备的纳米粒子进行了物理表征;采用循环伏安(CV)、计时电流(CA)、计时电位(CP)和电池测试等方法对其进行了电化学表征。
1 实验
1.1 实验所用试剂
实验所用试剂均为分析纯:NaOH,98%,西陇化工;NaBH4,98%,天津光复化工;NiCl2·6 H2O,北京化工厂;HAuCl4·4 H2O,阿拉丁;正辛烷,阿拉丁;聚氧乙烯月桂醚(Brij-30),99.99%,阿拉丁;Nafion溶液(质量分数5%),阿拉丁。
1.2 催化剂的制备
本文中,Ni@Au/C催化剂是在反相微乳液体系中采用连续两步化学还原法所制备,制备过程如下:
1.3 工作电极的制备
工作电极采用玻碳电极(3 mm),使用前先用Al2O3浆液充分擦拭,然后在超纯水和乙醇中各超声洗涤3次,晾干备用。称取10 mg制备好的催化剂加入到0.95 mL乙醇和0.05 mL Nafion混合溶液中。超声分散30 min。然后移取5 μL均匀涂于玻碳电极表面,置于80℃干燥箱中干燥10 min后取出,备用。
1.4 催化剂物理表征
采用日本Rigaku公司的X射线衍射仪(D/max-2700)对纳米粒子的晶型结构进行表征,辐射源为CuKα靶,衍射波波长为0.154 056 nm。采用JEM-2100F型高分辨率透射电子显微镜(TEM)对所制备纳米粒子的形貌和粒径进行表征,加速电压为200 kV。
1.5 催化剂电化学表征
采用美国PAR公司的VMPⅢ型多通道恒电位仪进行测试,电化学测试采用三电极体系,工作电极为负载着催化剂的玻碳电极,对电极为Pt网(1 cm×1 cm),参比电极为Hg/HgO (1 mol/L NaOH)。电池测试在自制的电化学槽中进行,采用三电极体系,阳极电解液:0.5 mol/L NaBH4+2 mol/L NaOH;阴极电解液:4.5 mol/L H2O2+2.0 mol/L HCl。阳极室和阴极室的内表面尺寸分别为 3 cm×4 cm×5 cm。电解液隔膜为NRE-117 Na+膜,暴露面大小为1 cm×1 cm。
2 结果与讨论
2.1 催化剂的物理表征
2.1.1 XRD表征结果
图1为Au/C和Ni@Au/C的XRD谱图。由图可知,Ni@Au/C表现出和Au/C相似的图谱,但并没有出现Ni的特征衍射峰,这可能是由于Ni核太小,衍射峰不明显引起的[6]。图中2 θ角位于38.2°、44.3°、64.7°、77.4°和81.7°的衍射峰分别对应面心立方(111)、(200)、(220)、(311)和(222)晶面Au的衍射峰。2 θ角位于24°的衍射峰对应于XC-72炭黑(002)晶面的特征衍射峰。从图1中还可看出,Ni@Au/C的(220)晶面的衍射峰较Au/C轻微地向左偏移,表明Ni@Au/C的晶胞较Au的要稍微大一些[7]。根据(220)衍射峰由公式(2)可算出Au/C和Ni@Au/C的晶粒大小分别为16.66和11.17 nm。
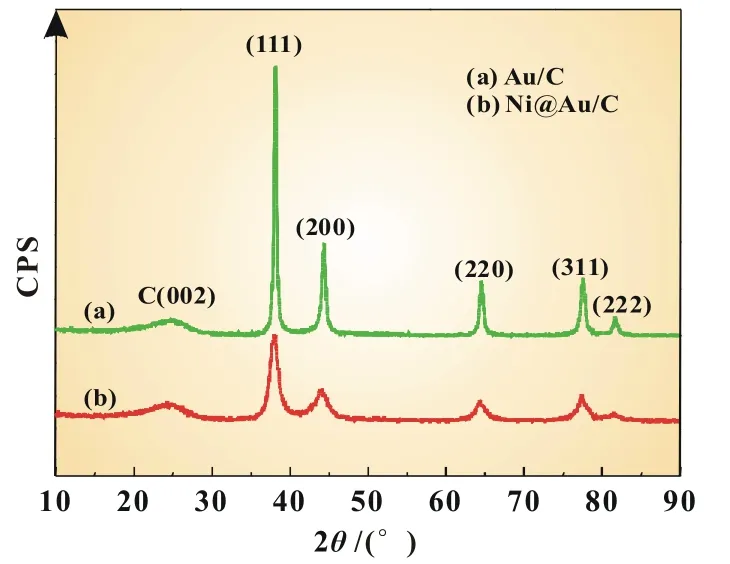
图1 Au/C和Ni@Au/C催化剂的XRD谱图

2.1.2 TEM表征结果
图2为Ni@Au/C纳米粒子的TEM照片。从图2(a)中可看出,所制备的纳米粒子近似为球形且比较均匀地分散在XC-72C表面。在图2(b)中,颜色较深的部分为Ni核,外层较浅的部分为Au壳层,表明所制备的Ni-Au纳米粒子为核壳结构。本文统计了大约100个纳米粒子,计算出Ni@Au/C的平均粒径为10 nm,与XRD得到的结果一致。
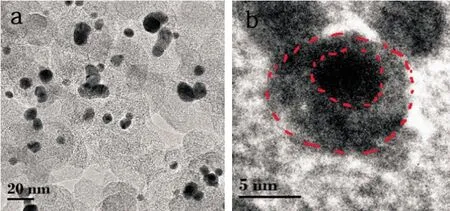
图2 Ni@Au/C纳米粒子的TEM照片
2.2 催化剂的电化学表征
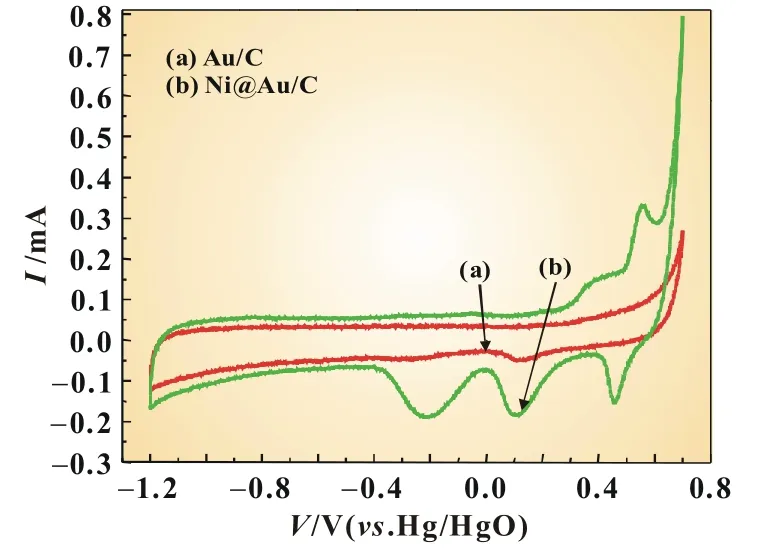
图3 Au/C和Ni@Au/C催化剂在氩气饱和的1.0 mol/L NaOH溶液中的循环伏安曲线
2.2.1 电化学活性面积的计算
催化剂的电化学活性面积(ECSA)可以通过循环伏安曲线进行计算。图3是Au/C和Ni@Au/C催化剂在氩气饱和的1.0 mol/L NaOH中的循环伏安曲线,扫描速率为50 mV/s,图中位于0.0~0.2 V的还原峰对应于氧原子在Au原子表面的氧化析出峰[8],单层氧原子从Au表面析出所需的电量为420 μC/cm2[9],因此,根据式(3)可计算出Au/C和Ni@Au/C的电化学活性面积分别为161.0和815.7 cm2/mg。
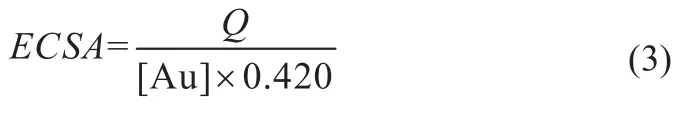
可以看出核壳结构的Ni@Au/C的ECSA约是Au/C的5倍,在同等条件下可能有更好的催化性能。
2.2.2 循环伏安测试
图4为两种催化剂在0.03 mol/L NaBH4+1.0 mol/L NaOH溶液中的循环伏安曲线,扫描范围:-1.2~0.6 V,扫描速率为50 mV/s,温度293 K。由图可知,Ni@Au/C催化剂和Au/C的CV曲线形状相似,表明反应机理相似。正向扫描时,Au/C和Ni@Au/C的CV曲线主要由位于-0.4~0.1 V的强氧化峰a1 (BH4-发生8 e-转移的直接电氧化反应而形成的[10])和0.4 V附近比较平坦的氧化峰a2(生成Au氧化物的范围[2])构成;逆向扫描时,出现了一个尖锐的还原峰c1(Au氧化物的还原),这和已报道的BH4-在Au/C上的循环伏安曲线形状基本一致[10-11]。从图4中还可看出,Ni@Au/C的a1氧化峰电位较Au/C负移了约0.2 V,氧化峰电位越负,表明BH4-的直接电氧化反应越容易发生。Ni@Au/C不仅有较负的氧化峰电位,而且能产生较大的电流密度(26.25 mA/cm2),用作DBFC阳极催化剂可能获得较好的性能。
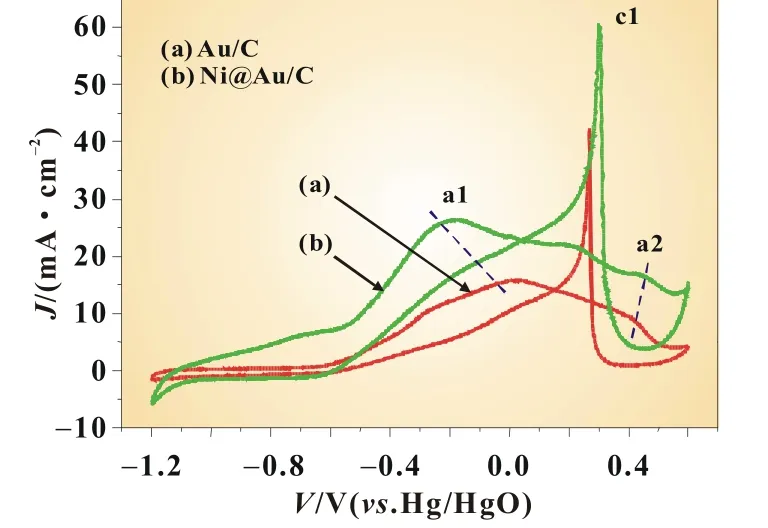
图4 Au/C和Ni@Au/C在0.03 mol/L NaBH4+1.0 mol/L NaOH中的循环伏安曲线
图5是以Au/C和Ni@Au/C作为阳极催化剂,在0.03 mol/L NaBH4+1.0 mol/L NaOH溶液中,当扫描速率从10 mV/s变化到750 mV/s时,氧化峰a1的峰值电流和扫描速率的平方根的关系图。由图可看出,pa1和1/2基本呈线性关系,表明BH4-在Au/C和Ni@Au/C催化剂上的电氧化过程受扩散控制,是一种典型的不可逆反应[12]。对于不可逆反应,满足Randles-Sevcik公式(4)和(5):
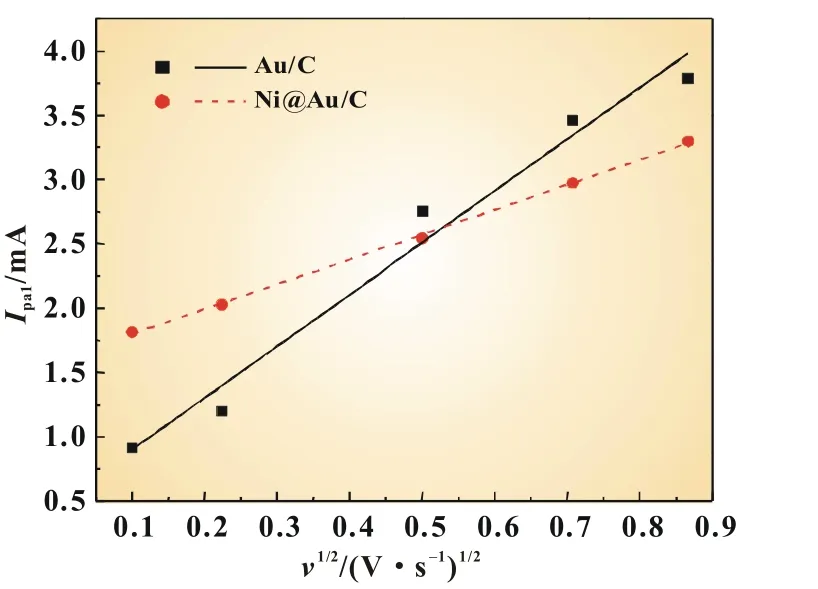
图5 氧化峰a1的峰电流(pa1)和扫描速率平方根(1/2)的关系
利用式(4)和(5)可计算出BH4-以Au/C和Ni@Au/C为催化剂,在a1峰附近转移的电子数分别为4.9和6.6,表明在BH4-电氧化过程中,Ni@Au/C比Au/C有更高的电子利用率。
2.2.3 计时电位和计时电流
图6为Au/C和Ni@Au/C催化剂在0.1 mol/L NaBH4+1.0 mol/L NaOH溶液中的计时电位曲线,给定恒定电流密度:8.5 mA/cm2。由图可知,在相同测试条件下,Ni@Au/C比Au/C有更低的开路电位,约为-1.14 V。测试 120 s后Au/C和Ni@Au/C的电极电位分别为-0.20和-0.79 V。比较可知Ni@Au/C有较低的过电位(0.28 V),较低的过电位预示着有较高的催化活性,发生氧化反应的电极电位越负,氧化反应越容易发生[14]。
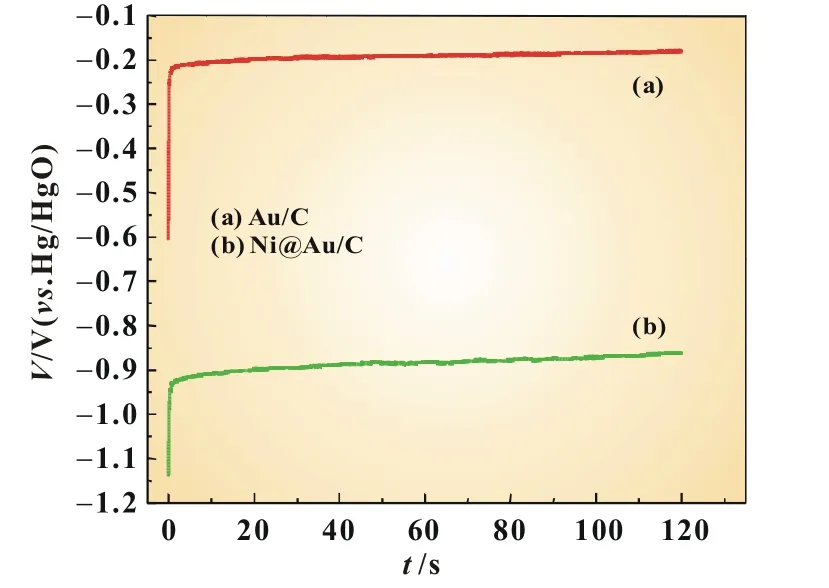
图6 Au/C和Ni@Au/C催化剂在0.1 mol/L NaBH4+1.0 mol/L NaOH中的计时电位曲线
图7为Au/C和Ni@Au/C催化剂在0.1 mol/L NaBH4+ 1.0 mol/L NaOH溶液中的计时电流曲线,阶跃电位:-1.2到-0.2 V。从图7中可以看出,测试开始时的前10 s,Au/C和Ni@Au/C对应的电流值迅速减小,直到20 s后电流值才基本趋于稳定;测试60 s后,催化剂Au/C和Ni@Au/C的电流值基本稳定,分别为7.4和54 mA/cm2。在相同测试条件下,核壳结构Ni@Au/C催化剂表现出比实心纳米Au/C更高的电流值,用作DBFC阳极催化剂时,可能会提供较大的输出电流。
2.2.4 单电池性能测试
图8是Au/C和Ni@Au/C催化剂在20℃时,用作DBHFC阳极催化剂时的极化曲线和功率密度曲线。从图中极化曲线可看出:Ni@Au/C催化剂的极化度最小,说明BH4-的电氧化反应在Ni@Au/C上更容易进行;在电流密度小于10 mA/cm2以前,电池电压呈线性骤降,当电流密度大于10 mA/cm2以后,电压下降速度减缓。这可能是由于在低过电势区域,极化过程为电极表面的吸附物种BH4-与OHad-进行电化学反应控制过程,此时电极反应可近似认为是零级反应,随着过电势的增大,电极过程逐渐转变为由BH4-化学吸附所控制的过程,产生极限电流,电极反应转变为一级反应[15]。本文中电流密度低于10 mA/cm2时,为电极表面极慢的电化学反应控制过程,电流密度大于10 mA/cm2时,电池电压随着电流密度的增加线性降低,可认为此时电池性能主要由电极欧姆极化所控制,整个实验过程中并没有出现极限电流,说明所制备的Au/C和Ni@Au/C催化剂具有优异的传质性能。
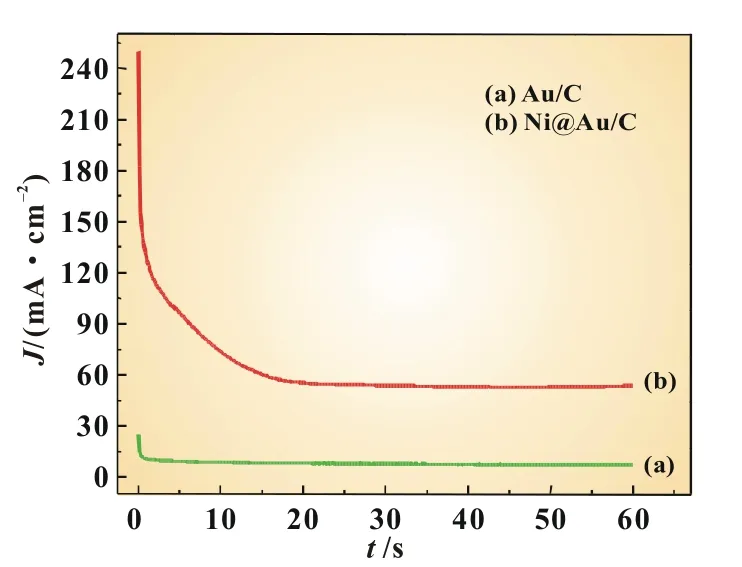
图7 Au/C和Ni@Au/C催化剂在0.1 mol/L NaBH4+1.0 mol/L NaOH中的计时电流曲线
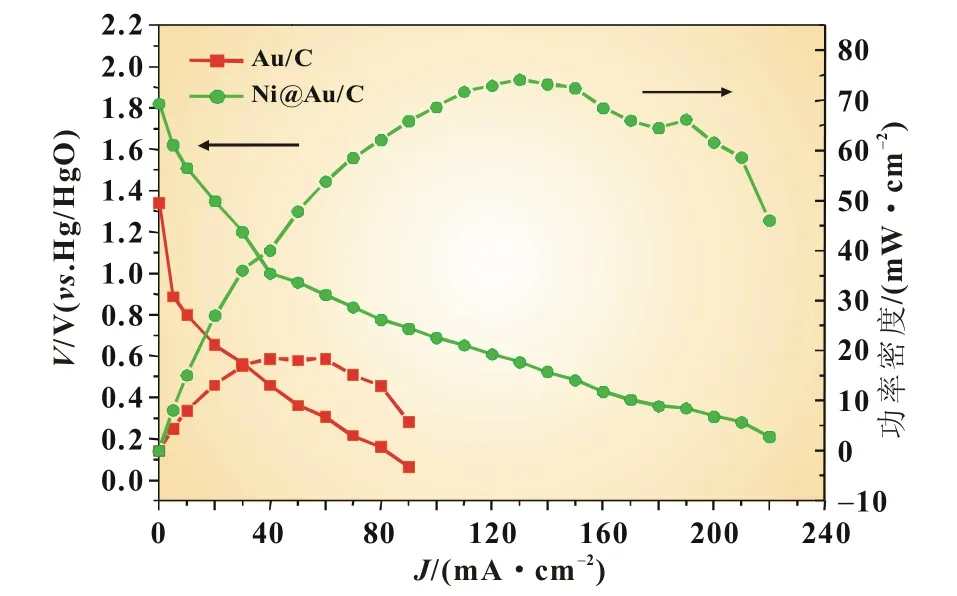
图8 Au/C和Ni@Au/C催化剂用作DBHFC阳极时的极化曲线和功率密度曲线
表1中列出了各催化剂的一些主要性能参数。由表1可知,Ni@Au/C催化剂有更高的开路电压(1.8 V),但是和理论电压(3.01 V)相比,仍然小了很多,这主要是由BH4-的中间产物氧化形成了混合电势引起的。Ni@Au/C催化剂在电流密度为130 mA/cm2时获得的最大功率密度为74 mW/cm2。可看出,用部分廉价金属Ni替代部分贵金属Au制成的核壳结构的Ni@Au/C催化剂的最大功率密度约是Au/C的4倍,因此,以Ni@Au/C作为DBFC阳极催化剂可能会取得较好的电池性能。

表1 Au/C和Ni@Au/C用作DBHFC阳极催化剂时的主要性能参数
3 结论
本文在反相微乳液体系中制备了碳载Au和核壳结构Ni@Au纳米粒子。物理表征表明,所制备的粒子为直径在10 nm左右的近似球形的核壳结构纳米粒子。电化学测试表明,Ni@Au/C纳米粒子有更大的电化学活性面积(815.7 cm2/mg),大约是Au/C的5倍,且BH4-在Ni@Au/C催化剂上的氧化反应较Au/C更容易发生,并且以a1氧化峰为基准,计算出BH4-在Au/C和Ni@Au/C上所转移的电子数分别为4.9和6.6。在20℃时,以负载着Ni@Au/C的玻碳电极为阳极电极,Pt网为阴极电极组装成DBHFC,获得的功率密度达到74 mW/cm2,约是相同条件下Au/C的4倍。因此,核壳结构Ni@Au/C是一种DBFC高效的阳极催化剂。
[1]岳增芳,余丹梅,陈昌国.硼氢化钠燃料电池负极催化剂的研究进展[J].电源技术,2011,35(1):113-115.
[2]CHATENET M,MICOUD F,ROCHE I,et al.Kinetics of sodium
borohydride direct oxidation and oxygen reduction in sodium hy
droxide electrolyte[J].Electrochim Acta,2006,51(25):5459-5467. [3]段东红,孙彦平.直接硼氢化物燃料电池(DBFC)阳极材料及反应机理[J].化学进展,2010,22(9):1720-1728.
[4]BEL N M C M,CHATENET M,LIMA F H B,et al.In situ fourier transform infrared spectroscopy and on-line differential electrochemical mass spectrometry study of the NH3BH3oxidation reaction on gold electrodes[J].Electrochim Acta,2013,89:607-615.
[5]HOSSEINI M G,ABDOLMALEKI M,ASHRAFPOOR S.Electrocatalytic oxidation of sodium borohydride on a nanoporous Ni/Zn-Ni electrode[J].Chinese J Catal,2012,33(11/12):1817-1824.
[6]SARKAR A,MANTHIRAM A.Synthesis of Pt@Cu core-shell nanoparticles by galvanic displacement of cu by Pt4+ions and their application as electrocatalysts for oxygen reduction reaction in fuel cells[J].J Phys Chem C,2010,115(10):4725-4732.
[7]LIU X,WANG A,LI L,et al.Structural changes of Au-Cu bimetallic catalysts in Co oxidation:In situ XRD,EPR,XANES,and FT-IR characterizations[J].J Catal,2011,278(2):288-296.
[8]TEGOU A,ARMYANOV S,VALOVA E,et al.Mixed platinum gold electrocatalysts for borohydride oxidation prepared by the galvanic replacement of nickel deposits[J].J Electroanal Chem,2009, 634(2):104-110.
[9]LIMA F H B,PASQUALETI A M,MOLINA C M B,et al.Borohydride electrooxidation on Au and Pt electrodes[J].Electrochim Acta, 2012,84:202-212.
[10]GYENGE E.Electrooxidation of borohydride on platinum and gold electrodes:Implications for direct borohydride fuel cells[J].Electrochim Acta,2004,49(6):965-978.
[11]CONCHA B M,CHATENET M,MAILLARD F,et al.In situ infrared(ftir)study of the mechanism of the borohydride oxidation reaction[J].PCCP,2010,12:11507-11516.
[12]NAGLE L C,ROHAN J F.Nanoporous gold anode catalyst for direct borohydride fuel cell[J].Int J Hydrogen Energy,2011,36(16): 10319-10326.
[13]DENUAULT G,MIRKIN M V,BARD A J.Direct determination of diffusion coefficients by chronoamperometry at microdisk elec trodes[J].J Electroanal Chem&Int Electrochem,1991,308(1/2): 27-38.
[14]YI L,LIU L,LIU X,et al.Carbon-supported Pt-Co nanoparticles as anode catalyst for direct borohydride-hydrogen peroxide fuel cell:Electrocatalysis and fuel cell performance[J].Int J Hydrogen Energy,2012,37(17):12650-12658.
[15]DUAN D,LIU S,SUN Y.Analysis of the kinetics of borohydride oxidation in Cu anode for direct borohydride fuel cell[J].J Power Sources,2012,210:198-203.
Studies of anode catalyst of sodium borohydride fuel cell
Carbon supported core-shell structure Ni-Au (Ni@Au/C)nanoparticles with the proportion of 1:1 was prepared by a two-step reduction method. The structure of the as-prepared catalysts was investigated by transmission electron microscopy(TEM)and X-ray diffraction (XRD),and the electro-oxidation activity towars to BH4-were tested by cyclic voltammetry(CV),chronopotentiometry(CP),chronoamperometry(CA)and fuel cell test. The results show as follows:the size of the core-shell structure nanoparticles is approximately 10 nm,and the electrochemically active surface area(ECSA),815.7 cm2/mg,is obtained,which is four times higher than that of Au/C catalyst.And a higher catalytic activity towards the electro-oxidation of BH4-wasexhibited of the Ni@Au/C catalyst. A single cell,which was fabricated using Ni@Au/C as anode catalysts and Pt mesh (1 cm×1 cm)as cathode electrode,achieved a maximum power density of 74 mW/cm2at 20℃.
core-shell structure nanoparticles;anode catalyst;nickel;golden;electro-oxidation;borohydride fuel cell
TM 911
A
1002-087 X(2015)10-2119-04

2015-03-11
山西省自然科学基金项目(2015011028);国家自然科学基金项目(20676088)
梁建伟(1987—),男,河北省人,硕士生,主要研究方向为燃料电池。
段东红,男,博士,副教授,E-mail:dhduan@163.com。
猜你喜欢
化工管理(2022年14期)2022-12-02
中学生数理化·中考版(2022年12期)2022-02-16
陶瓷学报(2021年1期)2021-04-13
中学生理科应试(2017年2期)2017-04-01
西安工程大学学报(2016年6期)2017-01-15
材料科学与工程学报(2016年1期)2017-01-15
材料科学与工程学报(2016年1期)2017-01-15
肇庆学院学报(2016年5期)2016-03-11
湖北师范大学学报(自然科学版)(2015年1期)2016-01-10
船舶标准化工程师(2015年5期)2015-12-03
