
利用半监督降维算法预测蛋白质亚细胞位置
2015-07-28 02:53:39薛建新谭文安上海第二工业大学计算机与信息工程学院上海201209
上海第二工业大学学报 2015年3期
王 彤,薛建新,谭文安(上海第二工业大学计算机与信息工程学院,上海201209)
利用半监督降维算法预测蛋白质亚细胞位置
王彤,薛建新,谭文安
(上海第二工业大学计算机与信息工程学院,上海201209)
摘要:首先采用伪氨基酸组成(PseAA)和特定位点记分矩阵(PSSM)2种方法组合的特征提取方法来表达蛋白质序列。通过该方法将蛋白质序列转化成特征向量,虽然该向量在很大程度上保留了蛋白质序列的原始信息,但是它产生的相应的维数会很高,这使得蛋白质亚细胞位置的预测过程变得很复杂。同时,就目前的情况来看,想要获取大量已标记的蛋白质亚细胞位置样本也很困难。为了解决这些问题,提出采用半监督降维算法(SS-MVP)对特征向量进行降维的同时能从标记和未标记的样本点中提取对分类有用的信息。基于降维后的样本利用支持向量机(SVM)的算法来预测蛋白质亚细胞位置类型。实验结果表明,采用上述方法既能简化蛋白质亚细胞位置的预测系统,又能提高其分类性能。
关键词:蛋白质亚细胞定位;预测;半监督;降维
0 引言
蛋白质亚细胞位置是指蛋白质所在的亚细胞[1]。人类正常的生命活动都需要蛋白质的介入,但要想蛋白质行使其功能,其必须位于正确的亚细胞位置才行,否则就会出现机体功能紊乱,甚至会产生疾病。因此,蛋白质亚细胞位置是蛋白质的关键特征之一[1]。蛋白质的亚细胞定位的研究为在蛋白质上进行的一系列研究,包括其功能、其之间的相互作用等都有帮助,甚至可以为今后开发药物靶标等方面的研究提供参考信息。
蛋白质亚细胞位置通过实验方法测定,也有很多工作提出不同的预测方法。近几年生物数据快速增长,如果仍用传统的物理化学去测定这些海量的生物数据,势必会产生很多问题,例如成本太高、耗时太长等。为此,研究人员致力于探索能自动、快速地识别蛋白质亚细胞位置的预测算法。
最近几年,Shen等[2]提出基于伪氨基酸组成(Pseudo Amino Acid,PseAAC)以及OET-KNN(Optimized Evidence-Theoretic K-Nearest Neighbor classifiers)算法来预测蛋白质的亚细胞位置。PseAA是一种蛋白序列的特征提取方法,该方法生成的特征向量中包含了氨基酸的物理和化学等信息[3],因此其性能要比传统的氨基酸组成方法(Amino Acid Composition,AAC)好。文献研究表明:亚细胞器中同时出现的蛋白质序列相似度较高[4]。特定位点记分矩阵方法(Position-Specific Scoring Matrix,PSSM)[5]是一种既涉及了蛋白质序列的相似度又兼顾了蛋白质的进化信息的特征提取方法。本文提出一种PseAA和PSSM相组合特征提取方法表示蛋白质序列,通过该方法将蛋白质序列转化成特征向量,虽然该向量在很大程度上保留了蛋白质序列的原始信息,但是它产生的相应的维数会很高。同时在实际应用中,要获取大量已标记的蛋白质亚细胞位置样本相对困难。所以,在获得少量已标记样本时,需要利用大量未标记样本来改善学习性能以提高分类器效果,为此提出了半监督-最大方差映射方法(Semi-Supervised Maximum Variance Projections,SSMVP)[6]来解决这个问题。实验结果表明该方法能有效预测蛋白质亚细胞位置。
1 材料和方法
1.1数据集
数据集由Shen等[2]构建,并已在网上公布,可以通过网址http://www.csbio.sjtu.edu.cn/bioinf/Nuc-PLoc/Data.htm下载。该数据集包含9类亚细胞定位区域,共有714条蛋白质序列:99条染色质,22条异染色质,61条核膜,29条核基质,79条核孔复合体, 67条核斑点,307条核仁,37条核质和13条核体。
给定一个待查询蛋白质序列,为了预测它的亚细胞位置,首先需要做的是使用合适的算法来对它进行表达。本文提出一种PseAA和PSSM相组合的方法来表达蛋白质序列。下面简要介绍PseAA和PSSM算法。
1.2PseAA序列编码方法
2001年,Chou等[3]改进了原始的氨基酸组成方法,提出了伪氨基酸组成PseAA,可表示为

式中:X为···;Ai(i=1,2,···,20+λ为···,λ为···。
该方法先用氨基酸组成方法生成20维特征向量,然后根据氨基酸残基的疏水性和亲水性等计算得到λ维量。蛋白质序列总共被离散化为一个20+λ维向量。该数据集中,蛋白质长度最短为50,故λ的最大值为49。因此,通过采用PseAA算法,每条蛋白质生成一个20+49=69维的向量。
1.3PSSM序列编码方法
PSSM序列编码方法是对PseAA方法的一个补充,从另一个角度引入了蛋白质序列的进化信息,由此蛋白质序列可以表示为一个420维的矩阵,如下所示:

式中:PPSSM-420为···;¯Ai(i=1,2,···,20)为···; Sj(j=1,2,···,400)为···。
人体内有20种氨基酸,每一种都用一个字母表示,向量中的下标1,2,···,20分别对应于某一种氨基酸。前面20项组成表示PSSM矩阵每一列的平均分数,PSSM矩阵可以表示为[7]:
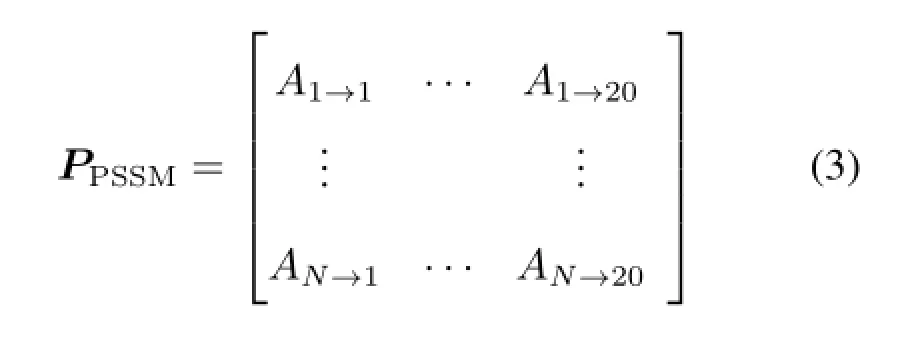
蛋白质P的长度为N,公式(3)中,分数Ai→j表示蛋白质序列P的第i个位置的氨基酸进化为第j种氨基酸时得到的分数[5]。下标1,2,···,20同样表示所对应的某种氨基酸[8]。
将PPSSM中对应于20种氨基酸的值分别求和,得到一个20×20的矩阵,即公式(2)中的S1,S2,···, S400。
本文采用PseAA算法和PSSM算法相组合的蛋白质特征表达方法,一条蛋白质序列最终可以被表示为一个69+420=489维的向量。该特征向量维数较高,会使蛋白质亚细胞位置预测问题复杂化,同时蛋白质亚细胞位置数据中存在了大量的未标记数据以及相对来说较少的有标记数据问题,为此提出了采用半监督降维算法SS-MVP来解决上述问题。
1.4SS-MVP半监督降维算法
数据集

是由m维的实数空间Rm内给出的,数据集包含C个类别[Φ1Φ2···ΦC],并且每个数据点xi(i=1,2,···,N)分别属于某一个类别。算法将原始数据X通过投影矩阵投影到低维空间Rd(d 新空间的数据集为 根据SS-MVP算法[6],投影变换矩阵A可通过最小化目标函数 得到,等价于最大化目标函数 其中:W=[wij](i,j=1,2,···,N)为系数矩阵, E=[eij](i,j=1,2,···,N)为相似度矩阵。想要最大化目标函数,即等价于最大化其分子和最小化其分母。 (1)最大化其分子,即 满足条件: (2)最小化其分母,即 如果xi和xj属于不同的类别,则wij=0,并且 算法计算的样本点都标记过类别信息,不包含未标记的样本子集 针对目标函数中的分子可以化简为: 式中:N=D−S,dii=;tr表示是对矩阵的对角线元素求和。 分母化简为: 式中,M=(I−W)T(I−W),I是单位阵。考虑到如下映射关系: 那么,目标函数可以转化为 然而样本数小于维数时就会产生奇异值问题。为了解决样本奇异问题,本文采用加入正则项的方法,令加入的正则项为R(α),则目标函数可以变为[9] 设G=(V,B)为邻接图,V是图G的结点集, B是边(xi,xj)的集合,(xi,xj)表示两相邻结点。邻接矩阵的定义为 定义正则项为 表1 不同蛋白质亚细胞定位算法的Jackknife预测结果比较Tab.1 The compasition of jackknife prediction results for different proteins subcelluler localization methods 通过推导(推导过程同式(5)),得到式中:L的含义类似于式(5)中N的含义。将R(α)的结果代入目标函数后,得到 转化为广义特征值求解问题[8]: 计算式(11)可得A,即子空间的投影转换矩阵。算法引入拉普拉斯矩阵的正则项[10],有助于从样本点(包含已标记和未标记)中提取对分类有用的信息,改善算法的分类性能。 数据集中包含714个蛋白质亚细胞位置数据,采用组合特征提取的方法提取其特征,应用SS-MVP算法对其降维,并将降维得到的714个数据利用SVM[11]分类器采用Jackknife测试方法进行Jackknife测试。表1、表2和表3所示为蛋白质亚细胞位置预测的实验结果。由于SVM只适用于解决数据类型有2种的分类问题,而本文要解决的蛋白质亚细胞位置类型预测问题包含多种类别,为此采用一对多方法将多类情况转化为多个2类问题,同时核函数采用多项式。 表2 每类蛋白质亚细胞位置降维前后的Jackknife测试准确率比较结果Tab.2 The Jackknife success rates for each of the subcelluler localization of proteins before and after dimensionality reduction 分别采用有监督降维算法MVP[12]和半监督降维算法SS-MVP预测蛋白质亚细胞定位数据的实验结果如表1所示。由表1可见,采用SS-MVP降维算法预测蛋白质亚细胞位置类型的Jackknife测试准确率是84%,高于采用MVP降维方法时的预测准确率。这就说明SS-MVP考虑了未标记样本点的类别信息。 表2是针对每种蛋白质亚细胞定位数据在降维前后的预测结果。由表2可见,采用SS-MVP降维算法降维前后,样本的特征向量分别为489维和60维,Jackknife测试的总的准确率分别为61%和84%。结果说明本文采用的SS-MVP降维算法不但解决了特征向量的“维数灾难”问题,同时还能提高预测的准确率[8]。 表3列出了分别采用3种不同的序列编码表达的蛋白质序列降维前后的Jackknife测试准确率比较结果。由表3可见,降维前,采用PseAA和PSSM的组合编码方法所得的预测准确率为61%,高于分别采用PseAA和PSSM编码方法的预测准确率;降维后,采用组合编码方法得到的预测准确率同样高于其他两种编码方法的预测准确率[7]。由此可见,使用组合编码方式及SS-MVP降维算法的预测准确率较高。 表3 采用不同的序列编码方法在原始高维向量和经过SS-MVP约简后的60维向量上辨识蛋白质的亚细胞定位Jackknife测试结果比较结果Tab.3 The Jackknife success rates for subcelluler localization of proteins based on original high dimensional vector and dimension-reduced 60-D vector generated by SS-MVP with three sequence encoding schemes 图1显示采用3种序列编码方法针对蛋白质特征向量降维后不同维数的Jackknife预测准确率比较结果。 由表1可见,降维后的特征向量维数范围在10 到100这个区间内,采用PseAA和PSSM的组合编码方法的预测准确率都高于分别采用PseAA和PSSM编码方法的预测准确率。同时,无论采用哪种序列编码方法,样本的特征向量在降到60维时的预测准确率都是最高的,所以实验的预测结果都采用特征向量为60维时的预测准确率。 通过采用半监督降维算法对特征向量进行降维的同时,还能从标记和未标记的样本点中提取对分类有用的信息。实验结果表明,采用上述方法既能简化蛋白质亚细胞位置的预测系统,又能提高其分类性能。 图1 采用3种序列编码方法针对蛋白质特征向量降维后不同维数的Jackknife预测准确率比较结果Fig.1 A plot to show the different overall jackknife success rates with various protein reduced vector dimensions generated by SS-MVP algorithm for three sequence encoding schemes 本文首先采用组合编码的特征提取方法来获取蛋白质序列的特征向量。然后采用半监督降维算法SS-MVP对该向量进行降维,并利用SVM来预测蛋白质亚细胞位置的类型。最后对降维的结果进行了Jackknife测试。实验结果表明,采用该方法降低了预测的复杂性,分类预测的准确率也得到了提升。 参考文献: [1]陈铭.生物信息学[M].北京:科学出版社,2012. [2]SHEN H B,CHOU K C.Nuc-PLoc:A new web-server for predictingproteinsubnuclearlocalizationbyfusingPseAA compositionandPsePSSM[J].ProteinEngineeringDesign &Selection,2007,20:561-567. [3]CHOU K C,CAI Y D.Predicting protein quaternary structure by pseudo amino acid composition[J].Proteins,2003, 53:282-289. [4]川韩榕.细胞生物学[M].北京:科学出版社,2011. [5]SCHAFFER A A,ARAVIND L,MADDEN T L,et al. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements[J].Nucleic Acids Research,2001,29:2994-3005. [6]WANG T,LU H,CAO X X,et al.Predicting protein subcellular localization based on a semi-supervised algorithm[C]//2012 International Symposium on Instrumen-tation and Measurement,Sensor Network and Automation(IMSNA).Sanya:IEEE Computer Society,2012:130-133. [7]王彤,杨志珍,曹晓夏.基于线性降维方法的蛋白质四级结构类型预测[J].上海第二工业大学学报,2013,30(1): 12-17. [8]王彤.高维生物数据的分类与预测研究[D].上海:上海交通大学,2009. [9]刘志宇.一种半监督判别邻域嵌入算法[J].计算机工程与应用,2011,47(19):173-175. [10]BELKIN M,NIYOGI P,SINDHWANI V.Manifold regularization:A geometric framework for learning from labeled and unlabeled examples[J].Journal of Machine Learning Research,2006,7:2399-2434. [11]HUA S J,SUN Z R.Support vector machine approach for protein subcellular localization prediction[J].Bioinformatics,2001,17:721-728. [12]ZHANG T H,YANG J,WANG H H,et al.Maximum variance projections for face recognition[J].Optical Engineering,2007,46:067206-1-067206-8. 中图分类号:TP391;Q617 文献标志码:A 文章编号:1001-4543(2015)03-0260-06 收稿日期:2015-03-10 通讯作者:王彤(1981–),女,山西人,副教授,博士,主要研究方向为数据挖掘、生物信息处理。电子邮箱wangtong@sspu.edu.cn。 基金项目:国家自然科学基金(No.61301249,No.61272036)、上海市自然科学基金(No.15ZR1417000)、上海市教委优青项目(No.ZZegd14001)、上海第二工业大学校基金(No.EGD14XQD13)资助 The Prediction of Protein Subcelluler Localization by Using Semi-Supervised Dimensionality Reduction Method WANG Tong,XUE Jian-xin,TAN Wen-an Abstract:Firstly,a fusion feature extraction method by combining Pseudo Amino Acid composition(PseAA)and Position-Specific Scoring Matrix(PSSM)is adopted to represent the features of proteins.Through this method,proteins are changed to feature vectors which can mostly retain the original information of protein sequence.But this high-dimensional feature vectors produced by using this fusion method may make the prediction system of protein subcelluler localization complex.At the same time,to obtain a large sample of marked protein subcellular location is also very difficult.To overcome these problems,a dimensionality reduction algorithm called Semi-Supervised Maximum Variance Projections(SS-MVP)is introduced to reduce the dimensional of feature vectors and extract useful information for classification from labeled and unlabeled sample points at the same time.Based on the reduced samples,Support Vector Machine(SVM)was applied for the prediction of protein subcelluler localization.Finally,the obtained results prove that the prediction system of protein subcelluler localization is simplified and classification performances are improved by adopting aboved methods. Keywords:subcelluler localization of protein;prediction;semi-supervised;dimension reduction

















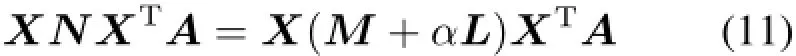
2 结果与讨论



3 结论
(School of Computer and Information Engineering,Shanghai Second Polytechnic University, Shanghai 201209,P.R.China)
猜你喜欢
黄河之声(2022年10期)2022-09-27 13:59:46
车主之友(2022年4期)2022-08-27 00:57:12
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25 13:08:00
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25 13:08:00
中学生数理化·高二版(2022年4期)2022-05-09 13:18:43
World Journal of Gastroenterology(2020年26期)2020-08-17 07:10:26
海峡姐妹(2019年12期)2020-01-14 03:24:40
疯狂英语·初中天地(2018年6期)2018-11-24 02:39:26
中学生数理化·八年级物理人教版(2017年11期)2017-04-18 11:22:51
火控雷达技术(2016年1期)2016-02-06 02:17:56
