
含N—乙酰化肝素寡糖的制备及序列分析
2015-07-09 10:27林江慧张建伟肖晓毛高建平张惠芳魏峥
分析化学 2015年5期
林江慧 张建伟 肖晓毛 高建平 张惠芳 魏峥


摘 要 建立了含N-乙酰化肝素寡糖的分离提纯及其序列结构分析方法。首先应用肝素酶Ⅰ深度酶解低分子量肝素来富集含N-乙酰化结构寡糖,通过Bio-Gel P10凝胶色谱法分离制备了包括二糖至十四糖的系列肝素寡糖粗样品,ProPac PA-1强阴离子高效液相色谱(SAX-HPLC)等方法对粗样品进一步分离,提纯得到4种六糖和3种八糖片段。其次应用肝素酶Ⅰ, Ⅱ和Ⅲ复合酶解与 HPLC法分析各纯化寡糖的二糖组分,并结合肝素酶Ⅰ底物特异性,初步推断4种六糖和3种八糖的序列结构。在寡糖的糖链两端均含有N-硫酸化二糖,而N-乙酰化二糖分布在糖链当中。应用电喷雾离子阱-飞行时间质谱(ESI-IT-TOF-MS)在负离子模式下进一步表征寡糖并分析其裂解规律。结果表明,各寡糖中均出现大量因SO2
3丢失形成的碎片离子峰,六糖中主要有双电荷和三电荷碎片离子峰;在八糖中出现了一系列从双电荷至五电荷的离子峰。各寡糖的双电荷离子峰质荷比进一步确定了上述寡糖的序列结构。六糖的裂解规律表明,裂解主要存在于糖苷键,N-乙酰葡糖胺和糖醛酸上的裂解方式分别为0,2X和0,2Z。本研究提供了切实有效的分离、分析未知结构肝素寡糖序列的新方法。
关键词 肝素寡糖; 飞行时间质谱; 肝素酶; 序列测定
1 引 言
肝素(Heparin, HP)是带有大量负电荷的线性糖胺聚糖,在细胞表面和细胞基质中广泛表达。HP与众多的蛋白质相互作用,调控多种生物学功能。除了抗凝血及抗血拴生成外, 肝素还具有抗平滑肌细胞增殖、抗炎症、抗肿瘤及抗病毒等, 而这些生物活性与肝素的特异结构密切相关【1~3】。
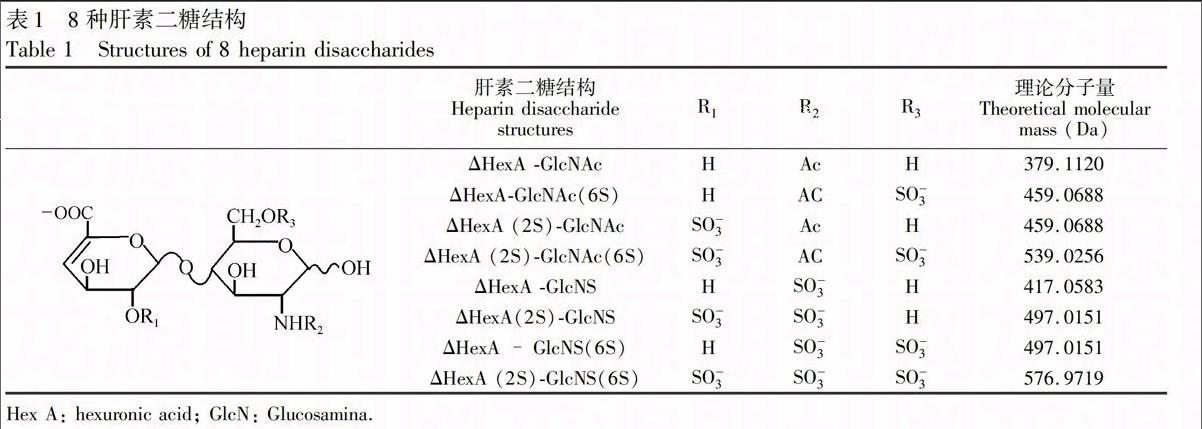
肝素聚糖链由50~200个重复的二糖单元构成。HP二糖是由己糖醛酸(Hexuronic acid, Hex A)与葡萄糖胺(Glucosamine, GlcN)以1-4糖苷键连接而成。己糖醛酸有两种结构,即葡萄糖醛酸或艾杜糖醛酸。己糖醛酸上的修饰有2位氧硫酸化,葡萄糖胺上的修饰有N位硫酸化、 N位乙酰化、C6位氧硫酸化,以及一些稀有结构 C3位硫酸化。这些修饰的组合构成了最常见的8种二糖单元(如表1所示)【4,5】。迄今为止评价HP结构主要是通过分析其酶解二糖组分,对功能性寡糖的序列研究报道不多。
高效液相色谱、凝胶色谱、毛细管电泳、聚丙烯酰胺凝胶电泳等技术已被应用于HP结构的研究【6】。NMR虽然能够测定肝素寡糖结构,但是需要样品纯度高且量较大,这限制了它在微量结构寡糖分析中的应用。由于质谱精度高,可快速分析糖类精细结构。近年来,质谱和液相色谱-质谱联用技术被大量应用于分析HP二糖或寡糖。Kailemia等应用质谱表征了一个类似肝素结构的五糖药物【7】。Huang等通过化学修饰,全序列分析了合成的肝素、硫酸类肝素寡糖类似物【8】。Huang等应用质谱电子转移裂解技术分析了硫酸类肝素寡糖【9】。Shi等应用串联质谱分析了肝素/硫酸类肝素寡糖的SO2
4丢失【10】。Kailemia等应用串联质谱分析了酶化学合成的肝素结构【11】。
肝素大部分结构是高硫结构,N-乙酰化结构含量较低,但这部分结构对于肝素功能也是至关重要的【11】。肝素六糖和八糖通过特殊的序列与多种蛋白质、酶等相互作用调控肝素的生理功能。肝素的抗凝血和抗带状疱疹病毒作用的六糖片段中C3位硫酸化和N位乙酰化部位是活性的必需位点【12】。肝素的N位基团是它与多种细胞增长因子相互结合的必要位点【13,14】,并且具有抑制肿瘤细胞来源的类肝素酶的活性【15】。最引人关注的是,低分子量肝素在临床上有直接的抗癌活性,它有助于延长肿瘤患者的生存时间,推测这与肝素的微量结构有着密切关系【16,17】。
分析微量结构肝素寡糖的序列,不仅对其结构与功能的研究具有重要意义,同时也为肝素作为有效的抑制剂提供了理论基础。肝素酶是制备二糖和寡糖的重要工具酶。其中肝素酶I对含有N位和C2位SO2
4的高硫二糖具有底物特异性【6】,对含N-乙酰化低硫结构不酶解。
本研究基于肝素酶Ⅰ底物特异性,采用二糖组分分析法,酶解低分子量肝素,分离制备含N-乙酰化肝素寡糖,初步确定了它们的序列结构,并通过质谱测定寡糖的质荷比及裂解规律。与文献报道中用质谱表征合成的已知结构的肝素寡糖类似物相比,本研究采用天然肝素对其寡糖的研究更具现实意义。
2 实验部分
2.1 仪器与试剂
1200系列高效液相色谱仪(HPLC,美国Agilent公司);Christ Alpha 1-4 LD Plus冷冻干燥机、Christ RVC 2-18真空离心浓缩仪(德国Christ公司); MS-IT-TOF离子阱飞行时间质谱、2450型紫外分光光度计 (日本岛津公司)。ProPac PA-1强阴离子交换(SAX)色谱柱(250 mm×4.0 mm,美国Dionex公司);Bio-Gel P10凝胶色谱柱 (115 cm × 1.7 cm,fine grade,美国Bio-Rad公司)。
低分子量肝素(分子量1000~14000 Da,英国Leo Laboratories公司);8种肝素标准二糖(ΔHexA(2S)-GlcNAc,ΔHexA-GlcNAc,ΔHexA-GlcNAc(6S),ΔHexA(2S)-GlcNS,ΔHexA-GlcNS,ΔHexA-GlcNS(6S),ΔHexA(2S)-GlcNAc(6S),ΔHexA(2S)-GlcNS(6S)),四糖(dp4),六糖(dp6),八糖(dp8),十糖(dp10),十二糖(dp12), 十四糖(dp14) (英国Idroun公司);肝素酶Ⅰ,肝素酶Ⅱ, 肝素酶 Ⅲ(美国Sigma公司);乙腈(色谱纯,德国Merck KGaA 公司);乙酸铵,碳酸氢铵(分析纯,美国Sigma公司);NaCl(色谱纯,美国Sigma公司);BSA(生物级,美国MPBio公司);其它试剂为国产分析纯。endprint
2.2 低分子量肝素深度酶解
200 mg低分子量肝素溶解在2 mL 0.1 mol/L乙酸钠缓冲溶液(含0.1 mmol/L乙酸钙和100 μg/mL BSA, pH 7.0)中,加入50 mIU肝素酶Ⅰ在37 ℃酶解12 h后,加入50 mIU肝素酶Ⅰ继续酶解12 h。加热至沸3 min终止反应。10000 r/min离心10 min,取上清液,冻干。
2.3 肝素寡糖的粗分离制备
上述样品用1 mL 0.2 mol/L NH4HCO3溶解,上样到Bio-Gel P10凝胶色谱层析柱(115 cm×1.7 cm)。0.2 mol/L NH4HCO3洗脱,流速0.2 mL/min。紫外检测波长232 nm,依次收集每个色谱峰对应的糖片段。55 ℃加热24 h 挥发NH4HCO3,冻干获得肝素寡糖粗样品。
2.4 肝素寡糖的细分与制备
ProPac PA-1柱强阴离子交换HPLC(250 mm×4.0 mm)进一步分离2.3节中获得的dp6和dp8寡糖。流动相A 为超纯水(以 HCl调至pH 3.5),流动相B 为, 2 mol/l NaCl(以 HCl调至pH 3.5)。dp6和dp8粗样品溶解在1 mL水(pH 3.5)中,上样。流速1 mL/min,线性梯度洗脱:0~2 min, 100% A; 2.1~7.1 min, 100%~60% A; 7.1~47.1 min, 60%~25% A。检测波长232 nm。收集色谱峰对应的寡糖样品,截留分子量500透析膜(美国Spectrum公司)透析2天,冻干,得到 dp6a, dp6b, dp6c, dp6d, dp8a, dp8b和dp8c 纯品。
2.5 肝素寡糖组分分析
2.5.1 各粗样组分分析 取2.3节制备的四糖(dp4)、六糖(dp6)、八糖(dp8)、十糖(dp10)、十二糖(dp12)、十四糖(dp14)、大于十四糖(>dp14)各50 μg,分别加入10 mIU肝素酶Ⅰ、Ⅱ、Ⅲ完全酶解,SAX HPLC分离。色谱条件如下:A 为超纯水(以 HCl调至pH 3.5),流动相B 为 2 mol/L NaCl(以HCl调至pH 3.5)。样品溶解于1 mL A中。色谱流速1 mL/min,线性梯度洗脱:0~2 min, 100% A;2.1~35.1 min,100%~75% A;35.1~57.1 min,75%~50% A。232 nm紫外检测。
2.5.2 寡糖dp6a, dp6b, dp6c, dp6d, dp8a, dp8b和dp8c 纯样品组分分析
取2.4节制备的dp6a , dp6b, dp6c, dp6d, dp8a , dp8b和dp8c各100 μg, 分别加入5 mIU的肝素酶Ⅰ, Ⅱ, Ⅲ完全酶解,SAX HPLC分离,色谱分离条件同上。
2.6 肝素寡糖质谱分析
离子化模式:ESI源;负离子模式;雾化气(高纯N2)流速1.50 L/min;干燥气(高纯N2)流速10 L/min;加热模块温度200 ℃;曲形脱溶剂管(CDL)
温度200 ℃;离子源电压4.5 kV;检测器电压1.6 kV;质量扫描范围:m/z 200~2000。准确吸取10 μL 50 μg/mL的dp6a, dp6b, dp6c, dp6d, dp8a, dp8b, dp8c,在负离子模式下,手动注入质谱仪。
图1 凝胶色谱柱分离肝素寡糖的色谱图
Fig.1 Separation of heparin oligosaccharides on
Bio-Gel P10 chromatographic column
洗脱液: 0.2 mol/L NH4HCO3,流速: 0.2 mL/min,紫外检测波长: 232 nm。
Elution: 0.2 mol/L NH4HCO3, flow rate: 0.2 mL/min, UV detection wavelength: 232 nm.
3 结果与讨论
3.1 肝素寡糖的粗分离及组分分析
如图1所示,肝素寡糖混合样品经Bio-Gel P10 凝胶层析柱,依据分子量大小分离,获得肝素二糖及一系列寡糖。通过与寡糖标样的洗脱时间对照(外标法)可知,图1中各峰对应二糖(dp2)、四糖(dp4)、六糖(dp6)、八糖(dp8)、十糖(dp10)、十二糖(dp12)、十四糖(dp14)以及 大于十四糖(>dp14)。从图1可见,Bio-GelP10凝胶色谱柱能够完全分离7种肝素寡糖和二糖,其中dp2的含量最高,随着寡糖分子量的增加,含量逐渐减少。由于肝素酶I对含有3个SO2
4的高硫二糖具有底物特异性【6】,不能酶解含N-乙酰化低硫二糖,因此推断以上系列寡糖中含有不同长度的N-乙酰化二糖结构。
2.3节中制备的寡糖dp4至>dp14按2.5.1节方法经肝素酶酶解-SAXHPLC分离,得到其二糖组分见表2,dp4至>dp14 的寡糖中均含有4种N-乙酰化二糖(dp4除外),4种N-硫酸化二糖。ΔHexA-GlcNAc随着寡糖分子量的增加,含量呈递增趋势。同样,ΔHexA-GlcNAc(6S) 和ΔHexA(2S)-GlcNAc的含量也呈现递增趋势。ΔHexA(2S)-GlcNAc(6S)含量变化不太。dp6至>dp14 的系列寡糖中,含N-乙酰化总二糖含量明显递增。上述结果表明,随着寡糖分子量的增加,N-乙酰化二糖增加的可能个数为1~3个。
N-硫酸化二糖含量变动趋势与N-乙酰化二糖不同。从dp6至>dp14 的寡糖中ΔHexA-GlcNS含量呈明显递增趋势,变动范围为1.9%→16.5%。ΔHexA-GlcNS(6S)呈递减趋势(20.8%→9.8%)。ΔHexA(2S)-GlcNS和ΔHexA(2S)-GlcNAc(6S)的含量变化不大,仅有3.0%和2.0%左右的波动。从表2可见,以>dp14为例,N-硫酸化二糖(NS-dp2)占总二糖含量为59.5%,2-O硫酸化二糖占43.5%,6-O硫酸化二糖(6S-dp2)占49.0%。寡糖系列中,随着分子量的增大,NS-dp2百分含量(84.3%→59.5%),2S-dp2百分含量(84%→43.5%)和 6S-dp2百分含量(87.0%→49.0%)都呈现降低的趋势。endprint
3.2 dp6,dp8分离提纯及序列分析
利用SAX-HPLC进一步分离dp6和dp8寡糖(见图2), dp6主要含有4个寡糖为dp6a, dp6b, dp6c, dp6d和dp8主要有3个寡糖dp8a, dp8b和dp8c。各提纯组分经肝素酶Ⅰ, Ⅱ和Ⅲ完全酶解后,SAX-HPLC分析,外标法鉴定其二糖组成及摩尔比(表2)。
表3列出了按2.5.2节中分析dp6和dp8 细分寡糖的组分及摩尔比。dp6a含有两种二糖结构ΔHexA-GlcNS, ΔHexA2S-GlcNS6S,摩尔比接近2∶1。基于肝素酶Ⅰ底物特异性,二糖GlcA2S-GlcNS6S在寡糖结构的末端。因此,dp6a序列结构为ΔHexA-GlcNS-HexA-GlcNS-HexA2S-GlcNS6S。按同理分析为了解质谱在测试寡糖中的精确度,表4列出了双电荷理论质荷比、实测质荷比及偏差。偏差范围在
34.1~37.4 ppm 之间。肝素寡糖质谱相对较复杂,质谱数据中还能观测到一系列的同位素峰。
References
1 Robinson C J,Stringer S E. J. Cell Sci., 2001, 114: 853-865
2 Couch man J R. Nat. Rev. Mol. Cell Biol., 2003, 4: 926-937
3 Kreuger J, Spillmann D, Li J P, Linda hl U. J. Cell Biol., 2006, 174: 323-327
4 Wei W, Ninonuevo M R, Sharma A, Danan-Leon L M, Leary J A. Anal. Chem., 2011, 83(10): 3703-3708
5 Yang B, Solakyildirim K, Chang Y, Linhardt R. J. Anal.Bioanal. Chem., 2011, 399(2): 541-557
6 Wei Zheng, Lyon M, Gallagher J T. J. Biol. Chem., 2005, 280: 15742-15748
7 Kailemia M J, Li L, Ly M, Linhardt RJ, Amster I J. Anal. Chem., 2012, 84(13): 5475-5478
8 Huang R, Liu J, Sharp J S. Anal. Chem., 2013, 85 (12): 5787-5795
9 Huang Y, Yu X, Mao Y, Costello C E, Zaia J, Lin C. Anal. Chem., 2013, 85(24): 11979-11986
10 Shi X, Huang Y, Mao Y, Naimy H, Zaia J. J. Am. Soc. Mass Spectrom., 2012, 23(9):1498-1511
11 Kailemia M J, Li L, Xu Y, Liu J, Linhardt R J, Amster I J. Mol. Cell Proteomics., 2013, 12(4): 979-990
12 Choudhary S, Marquez M, Alencastro F, Spors F, Zhao Y, Tiwari V. J. Biomed. Biotechnol., 2011, 2011: 264350
13 Wei Z, Deakin J A, Blaum B S, Uhrin D, Gallagher J T, Lyon M. Glycoconj. J, 2011, 28(8-9): 525-535
14 Catlow K R, Deakin J A, Wei Z, Delehedde M, Fernig D G, Gherardi E, Gallagher J T, Pavo M S G, Lyon M. J. Biol. Chem., 2008, 283: 5235-5238
15 Borsig L.Prog. Mol. Biol. Transl. Sci., 2010, 93: 335-349
16 Nicholas R L. J. Clin. Oncol., 2005, 23: 2119-2120
17 Agnes Y Y L, Frederick R R. J. Clin. Oncol., 2005, 23: 2123-2135
18 Zaia J, Costello C E. Anal. Chem., 2003, 75: 2445-2455
19 Wol J J, Laremore T N, Busch A M , Linhardt R J, Amster I J. J. Am. Soc. Mass Spectrom., 2008, 19: 790-798endprint
