
Analysis and Evaluation of Measures for the Administration of Medical Device Registration
2015-07-01 14:59:03LIFeiBIKaishunSchoolofBusinessAdministrationShenyangPharmaceuticalUniversityShenyang006ChinaLiaoningCenterforDrugandDeviceEvaluationandMonitoringShenyang0003China
亚洲社会药学杂志 2015年3期
LI Fei, BI Kai-shun(.School of Business Administration, Shenyang Pharmaceutical University, Shenyang 006, China; 2. Liaoning Center for Drug and Device Evaluation and Monitoring, Shenyang 0003, China)
Analysis and Evaluation of Measures for the Administration of Medical Device Registration
LI Fei1,2, BI Kai-shun1
(1.School of Business Administration, Shenyang Pharmaceutical University, Shenyang 110016, China; 2. Liaoning Center for Drug and Device Evaluation and Monitoring, Shenyang 110003, China)
Objective To study Measures for the Administration of Medical Device Registrations published by CFDA in 2014 and to summarize its impact on medical device market access in China. Methods Comparative study was adopted to conduct a research on the new and old measures for the administration, and systematic analysis was used to study the innovations of the measures for the administration. Results and Conclusion The new measures for the administration have made some improvements in the statutory code, efficiency and convenience as well as unity of licensing and supervision as an administrative license system. Additionally, the new provisions have made innovations on the subject qualification, evaluation principles and evaluation system. In order to implement the new measures for the administration, further reforms should be carried out on the uniform standards and personnel training to promote the registration management system.
medical device; measures for the administration of registration; medical device; market access; administrative license
1 Preface
“Regulations on Supervision and Administration of Medical Devices” came into operation on June 1st, 2014. As a higher level law, it can be implemented with related supporting rules. Therefore, China Food and Drug Administration has worked on the revision of rules related to“Regulations on Supervision and Administration of Medical Devices”. Among those rules, Provisions on Medical Device Registration (hereinafter referred to as “the Provision”) is the core regulation to standardize the registration of medical devices. The Provision was published on July 30th and put into force on October 1st, 2014.
The provision consists of 11 chapters with 82 articles, and it is divided into general principle, essential requirement, technical requirement & test, clinical evaluation, product registration, change of registration, registration prolongation, product filing for record, supervision and administration, legal responsibility and supplementary articles.
Among these chapters, essential requirement, technical requirement & test, clinical evaluation are requirements on market access, product registration, change of registration, registration prolongation and product filing for record are requirements for different forms of registration. Supervision and administration, and legal responsibility are restrains on registration activity.
2 Market access and registration types in the new provision
2.1 Changes of the market access
In terms of medical device market access, five main changes are listed in Table 1.
(1) Manufacturing license is not a requirement before registration.
This will help clarify relationships between registration certificate, manufacturing license and business license, which further defines that registration certificate of medical device is the certificate for market access. It is consistent with international medical device regulators. Premarketing reviews of medical device firms usually include quality system inspection (e.g. USA and EU) or manufacturing license review (e.g. Japan). In order to achieve our reform of national decentralization,manufacturing license should be managed in accordance with registration system.

Table 1 The main changes of market access between the new and old provision
(2) Product technical requirements substitute for the registered product standard.
Product technical requirements refer to the performance of medical devices and testing methods. Compared with the registered product standard, some of product specifications are deleted, but the product requirements and testing methods retain, which is the important basis for the production of medical devices. Therefore, product technical requirements can replace the registered product standard to achieve a smooth transition.
Product technical requirements, on the other hand, shall be approved by the Food and Drug Supervision and Administration Department. It is combined with the national standards and industry standards to work as the basis of registration, inspection and technical evaluation which gives it the legal status. Such provisions take the standard method as a proof of product rather than enforcement, and it is in line with the international standard.
(3) Substituting clinical evaluation for clinical trials and clinical validation.
Clinical evidence is made up of clinical evaluation report and the corresponding supporting documents; clinical evaluation is the clinical data. Clinical data come from several aspects, such as from the literature data, data from the same product in the clinical use and data from clinical trials. Therefore, clinical trial is part of the clinical data. International Medical Device Regulator Forum (IMDRF), Food and Drug Administration (FDA) and EU adopt this method to evaluate the product’s safety and effectiveness. Only when the product’s safety and effectiveness can’t be verified by the result of clinical literature and empirical data, clinical trial data must be provided. This method complies with ethical standard and it can prevent unnecessary trials. Meanwhile, clinical examination and approval is added to some high risk products such as implanted pacemaker, artificial heart valve. Whether to approve the clinical trial is based on the comparative analysis of risk degree, protocol and clinical benefit and risk.
(4) Adjusting the registration review time.
The review time for class II products has been prolonged from 40 working days to 60 days, and the review time for class Ⅲ products has been prolonged from 70 working days to 90 days. Furthermore, the new added clinical approval takes 60 working days and document amendment has been prolonged from 60 working days to one year. The extended document amendment time offers applicants plenty of time to do registration examination, clinical trials, which reduce the risk of returned review due to the overtime supplementary material.
(5) Registration certificate’s term of validity is increased from four years to five years.
After modification, the registration certificate’s term of validity is the same with manufacturing license and business license, and it is five years. Therefore, the applicants have enough time to manage their activities.
At the same time, the registered number remains the same which provide an opportunity to trace back the product in the whole lifecycle. It not only solves the problem of the invalid original packaging and labeling after the certificate expiration, but also provides traceable means of postmarketing surveillance.
2.2 Changes of the registration types
There are three types of medical device registration, the former “first time registration” was changed into“registration”, and the former “re-registration” and“registration change” were changed into “registration prolongation” and “the change of registration” respectively. As to the documents, registration documents are required, e.g. “the basic list of safety and effectiveness of medical device”, and it will help to set the requirements of registration scientifically.
The new prolongation registration gives fullconsideration to the interest of the applicants, but attention should be paid to three issues. Firstly, technical levels of the three requirements are different, particularly the second one demands a technical review which cannot simplify time limit and procedure. Secondly, the time limit of six months is mandatory. If the applicants miss the time for prolongation due to personal reasons, a site inspection or product testing, they have to apply for registration again. Thirdly, if decision from the regulator is overdue, it will be prolonged automatically. But it can be regarded as a decision of rejection when the regulator responsible for technical evaluation asks for supplementary documents and the applicants can’t demonstrate the products meet the need of requirement 2&3. At this situation, if the applicants can’t provide the required documents in six months or within the valid period, production can't be preceded until registration certificate is re-issued.

Table 2 Changes of registration types
3 The innovations of the new provision for registration and registration review
The new Provision has several innovations, such as using clinical evaluation and the registration prolongation, etc. But as a medical device management regulation, its core function is to regulate registration and review. The innovations are shown from three aspects: the qualification of the applicant for registration, the evaluation principles of registration and the systemic correlation of registration.
3.1 Qualification of the applicant
Qualification is the most concerned issue after the issuance of the “Regulations on Supervision and Administration of Medical Devices”. Registration license and production license of medical devices are the two cornerstones of medical device approval system. Licensees play an important role in licensing system, and it is vital to identify the relation of registration items with production items. The legal position of the licensee directly connects with the integrity, rationality and practicability of the licensing system of medical device.
The Provision defines that “The applicant for registration or filing should establish the quality control system related to product development and production and ensure its effective operation.” Hence, the licensee of registration and the licensee of production should be one subject, a legitimate enterprise which meets the requirements of license, marketing its medical devices by itself. It should be liable for the product and possess the registration certificate or archival filing certification, production license or archival filing certificate after the approval.
Innovative products have been given the support of exemption by the Provision, that is, when domestic enterprises apply for special approval of registration as innovative products, if the production of the sample is outsourced, the contractor should have the same production capacity. Hence, registrant of innovative medical devices can market his products more rapidly at a lower cost through outsourcing and the national innovation industries and the ordinary consumers will benefit from it greatly.
3.2 Evaluation principles of registration
According to the new Provision, medical devices registration refers to a process that the China Food and Drug Administration Department decides whether to approve a medical device application or not after conducting a systematic evaluation on the research of safety and effectiveness and result of the medical devices. From the definition, the core of registration is the evaluation of the safety and effectiveness of medical devices.
China implemented medical device registration system 20 years ago, and 14 years has passed since the implementation of “Regulations on Supervision and Administration of Medical Devices”, but China did not have the principles of registration. USA and European Union have accumulated abundant experience in medical device supervision based on years of research. They jointly set up the GHTF (Global Harmonization Task Force, predecessorof IMDRF (International Medical Device Regulators Forum), and finally issued the “Essential Principles of Safety and Performance of Medical Devices” and “Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices (STED)”.
“Essential Principles of Safety and Performance of Medical Devices (hereinafter referred to as EP)” clarifies the requirements for safety and performance of medical device in the product’s life cycle, which includes 59 articles. This is the evaluation principles of registration commonly used by most advanced countries and regions.
EP checklist is an acknowledged tool to implement 59 articles of EP. The checklist is composed of 3 parts, essential requirement, method used to verify conformity to essential requirement and evidences for verifying conformity to the essential requirement. In this way, the foundation of systematic evaluation is built. The checklist has been widely used by member countries of IMDRF. For instance, FDA requests “Declaration of Conformity and Summary Reports”in 510(k) files, EU requests “Essential Requirements Checklist” should be submitted in the application of medical device registration, and Japan has adopted STED (Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of Medical Devices) including “EP Checklist”.
The eleventh article of the newly issued Provision has formally clarified: the applicant should conform to EP when applying for registration. It is an important design of the new Provision, innovatively presenting EP as an evaluation principle.
3.3 The correlation of site inspection in registration
Medical device registration evaluation can be viewed as a system and its hierarchy is shown in Figure 1.
In the old Provision, site inspection is isolated from registration evaluation, which has potential risks. In most cases, site inspections are carried out by regulation personnel, and registration evaluation professionals are not involved. What is more, the inspection reports have conclusion without the inspection process and the control of R&D, thus there are two potential risks.
First, the application files are prepared only for registration, which are not real evidence controlled in quality management system (QMS). This makes registration detached from real conditions, and the registration cannot work as the permission for market access.
Secondly, in the process of the registration evaluation, the changes of product design, supplementary evidence and process adjustment (problems and risk control results initiated by evaluation) will affect the application files rather than the real QMS files. As a result, the registration process cannot control post-marketing risk.
The Provision requires the applicant must set up the quality management system related to product development and production so that the Food and Drug Administration Department can review the original research data in technical evaluation. Based on this, we can make two conclusions:
First, the product with registration certificate should be designed, produced and sold under the quality management system.
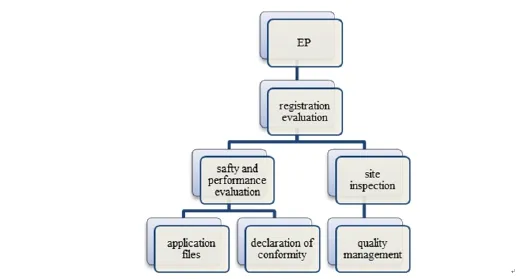
Figure 1 Registration evaluation system
Second, inspection of quality management systemshould be included in the systematic evaluation of medical device registration. Class II&III products should have onsite inspection before registration approval or at the same time.
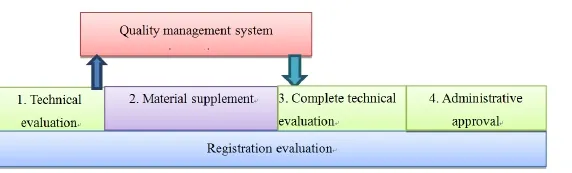
Figure 2 Inspection of quality management system in critical route
From the perspective of system analysis, the new provision offers a systematic guarantee for the combination of registration evaluation and site inspection. First, in the process of technical evaluation when the on-site inspection starts, the evaluation results should be used as part of the inspection scheme and key points. Second, the inspection results should not be used as permission; they should be used to confirm whether the raw materials, process, structure, composition and performance of the medical devices are consistent with application files. Thus they can work as the decision-making reference for registration evaluation. So a two-way feedback mechanism is formed, which can not only improve the effectiveness of on-site inspection, but also can promote the consistency of technical evaluation and the actual situation.
4 Evaluation of the provision based on the theory of administrative license
Administrative license is a relatively special legal system which is not only a premarket regulation method for government to control social risks, an allocation of social resources and improvement of the social public trust, but also a system to guarantee people to obtain specific rights and interests. Medical devices registration is an administrative license system carried out by the government to permit market access according to the risk of product. As an administrative license, the provision should follow the legal permission principle, the effectiveness and convenience principle, combined with supervision principle[4,5].
4.1 Based on the legal permission principle, the new provision has comprehensive regulations on application and evaluation
Based on the legal permission principle, the Provision has made a comprehensive design for current medical devices market access. It sets up a comprehensive regulations system, which uses the Provision as the core including notification, application file requirements, exemption catalogue, clinical evaluation principle and other normative documents, thus it can ensure that all aspects of the licensing items must follow the rules.
4.2 Based on the effectiveness and convenience principle, the new provision has improved the effectiveness of registration
Based on the legal permission principle, the Provision has clearly defined the time limit for each process of market access, the administrative organization shall, in accordance with the legal procedures within the prescribed time limit, handle the licensing matters without undue delay. At the same time, the Provision strengthens the regulation according to the registration risks, such as firsttime registration and clinical trial approval requirements. We can reduce the regulation adjustment by canceling unnecessary demands in the process of prolonging registration. Thus, the Provision regulates the market access with the minimum control cost (the shortest time, the least amount of manpower, financial and material resources and the least damage, etc.) to achieve the goal of administrative management, that is, to maximize social benefits.
4.3 Based on the principle of the combination of licensing and supervision, the new provision puts the on-site inspection and registration evaluation together
Based on the combined supervision principle, the old Provision did not reflect this principle. On the one hand, there is the risk that data in the application file are notconsistent with the actual condition, but the administrative department does not carry out supervision; on the other hand, the on-site inspection is lack of information support in the process of registration evaluation, and the risk is they do not have target for the on-site inspection.
For the first time the new Provision puts forward the idea that the original research documents can be used for the technical review. The applicant’s quality management system related to product development and production should be inspected as well. Therefore the approval of the registration license and the on-site supervision are combined to facilitate the efficiency of the administrative examination and approval, and the applicant will provide real application data voluntarily.
5 Problems and suggestions of the new provision
“Provisions on Medical Device Registration” is a code of conduct, and the subject is the regulators and applicants, so the regulators and applicants are the main factors influencing the implementation of the Provision. Most of the problems are caused by these people.
5.1 Taking measures to unify regulation standards and reduce the discretion
At present there is a phenomenon that various standards for registration evaluation exist. For example, different registration authorities usually made different evaluation results for the same kind of product in that they use various standards. In 2013, CFDA conducted a comprehensive screening on 361 medical device registration authorities and their approval of 22267 Class I medical device registration certificate data nationwide. As a result, CFDA found 2096 products with problems and took corrective measures for them, accounting for 9.4% of the total Class I products. The new Provision uses graded registration evaluation system which results in more than 300 registration authorities with approval power in China. Because there are still quality and ability differences among the personnel as well as the different approval scale, thus the results of registration evaluation are various. In the light of foreign experience, EU has carried out the graded examination and approval system, namely a third party implements the review scheme; there is also the problem of different scales review. Therefore, the classification management is the institutional reason to cause different evaluation scales.
This problem can be solved by taking uniform standard in advance and strengthening assessment methods afterwards. On the one hand, the guiding principle should be refined with detailed standards to reduce the discretion. On the other hand, an assessment mechanism should be established for the regulatory departments, especially the review agencies. Regular inspection and assessment of provincial medical device registration management should be carried out.
5.2 Strengthening registration evaluation personnel training and improving the efficiency of registration
In 2014, the number of Chinese medical devices product registration certificates was 26655, a growth of 14% than that in 2013. The number of applicants rose sharply which led to a big shortage of resources for registration evaluation. Take the technology review resources for example, the number of technical evaluation personnel at all levels was seriously insufficient with uneven professional structure, and the professional evaluation team should be set up urgently. There are more than 300 people engaged in medical device registration evaluation in FDA, and the European Union has 74 agencies for medical devices review, and 23 agencies for examining the in vitro diagnostic reagent. While China has 70 people engaged in medical device registration evaluation at the national level currently and 140 people engaged in medical device registration evaluation at the provincial level[6]. The annual per capita workload at the national level was 140 / person, and the annual per capita workload at the provincial level was 114 / person[7]. Based on the standard stipulated in “The guidance of strengthening the construction of provincial medical instrument technical evaluation ability” released by CFDA, the workload of national and local medical devices registration evaluation personnel was 3 times higher than that of the average workload. This led to some medical devices approval beyond the statutory time limit. Although some registration did not go beyond the statutory time limit due to the supplementary data, it took a long time in the administrative link, and there was a gap between administrative department and public expectation.
This problem can be solved by increasing the number of personnel, providing on-the-job education and performance evaluation incentive. First, a diversified review personnel mechanism should be established to form a team to meet the actual demand of the review gradually. Second, social resources such as experts from colleges, social organizations and healthcare organization should be used to help expand the ability and flexibility[8]. Third,strengthening the personnel assessment with in-service education, training and other incentives to fully mobilize the enthusiasm and initiative of staff.
In summary, as an administrative license system, the new Provision has made reform on the statutory code, efficiency and convenience as well as the unity of licensing and supervision. Additionally, the new Provision has made innovations on qualification, evaluation principles and system. In order to implement the new Provision, further reform should be carried out on the uniform standards, personnel training to improve the registration management system.
[1] LI Fei, YUAN Peng. The Application to GHTF Essential Principles of Safety and Performance for the Medical Devices Conformity Assessment in China [J]. China Medical Device Information, 2013, 18 (4): 1-8.
[2] YUAN Fu-qiang, LI Fei. Clinical Evaluation Research of Medical Device Registration [J]. China Medical Equipment, 2015, 12 (2): 34-37.
[3] YUAN Peng, LI Fei. Study on the New Submission Documents for Medical Device Registration [J]. China Medical Information, 2014, 19 (12): 5-8.
[4] YING Song-nian, YANG Xie-jun. The Theory and System Interpretation of the Administrative Permit Law [M]. Beijing: Peking University Press, 2004: 90-112.
[5] ZHOU You-yong. The Theory and Practice of the Administrative Permit Law [M]. Wuhan: Wuhan University Press, 2004: 41-52.
[6] WANG Lan-ming. Situation and Assessment on the Medical Device Registration System in China [J]. China Medical Information, 2012, 17 (11): 28-34.
[7] CFDA. Annual Report of Medical Device Registration (2014) [M]. Beijing: CFDA, 2014: 4-5.
[8] SUN Qin, YAN Liang. Better Medical Devices Regulations for Better Health Care: Enlightenment for Medical Devices Regulatory Reform in China, from Experiences of the EU and the USA [J]. Chinese Journal of Medical Instrumentation, 2006, 30 (1): 47-56.
Author’s information: BI Kai-shun, Professor. Major research area: National drug system. Tel: 024-23986012, E-mail: bikaishun@126.com

- 亚洲社会药学杂志的其它文章
- Risk Reviews of Preclinical Pharmaceutical Studies on Traditional Chinese Medicine Injection
- Risks and Risk Control Measures of in vivo Genotoxicity Test in New Drug R&D
- Regional Studies of Bio-pharmaceutical Industry in China
- GMP Regulations in China: Problems and Countermeasures
- Australian PBS and Its Enlightenment to Drug Price Reform in China
- Hot Issues about Drug Price in China from the View of Internet Public Opinion Monitoring