
Chediak-Higashi综合征的诊治探讨:一例报道并文献复习
2015-06-21 12:51深圳市儿童医院呼吸科广东深圳518026
罕少疾病杂志 2015年2期
1.深圳市儿童医院呼吸科 (广东 深圳 518026)
2.深圳市儿童医院血液肿瘤科 (广东 深圳 518026)
3.深圳市第六人民医院儿科 (广东 深圳 518052)
4.深圳市儿童医院检验科 (广东 深圳 518026)
陈杰华1李长钢2石红松2刘四喜2王 姝3徐 刚4
·胸部疾病·
Chediak-Higashi综合征的诊治探讨:一例报道并文献复习
1.深圳市儿童医院呼吸科 (广东 深圳 518026)
2.深圳市儿童医院血液肿瘤科 (广东 深圳 518026)
3.深圳市第六人民医院儿科 (广东 深圳 518052)
4.深圳市儿童医院检验科 (广东 深圳 518026)
陈杰华1李长钢2石红松2刘四喜2王 姝3徐 刚4
目的 探讨Chediak-Higashi综合征“加速期”的诊断与治疗。方法 报道1例Chediak-Higashi综合征并结合检索国内文献,采集病例资料,依据不同治疗方案或预后分组比较临床指标。结果 获得34篇文献,共计52例病例。男:女比例1.17∶1;25%患者有家族史;25.8%患者父母近亲结婚。92.3%患者因发热就诊,多有反复感染病史,97.9%皮肤或毛发色素异常;78.8%淋巴结增大,86.5%脾脏增大,86.5%肝脏增大。64.2%患者血常规两系以上异常。所有病例血涂片或骨髓检查可见白细胞胞浆内巨大包涵体。化疗组与未化疗组比较,治疗好转率(100% vs 50%,P =0.005)。治疗无效/死亡组与好转组比较,年龄(岁)(1.42±1.30 vs 3.61±1.81 P=0.0016),脾脏(cm)(6.1±3.4 vs 3.5±2.1,P=0.02),肝脏(cm)(4.3±2.4 vs 3.0±1.4,P=0.097),血常规两系降低比例(66.7% vs 55.6%,P=0.58)。结论 大部分CHS患者表现为反复发热,肝、脾、淋巴结肿大,血常规两系以上异常,不足以判断“加速期”。发病年龄可作为化疗的参考指征,婴儿期发病者死亡率高,化疗有利于控制病情。
Chediak-Higashi综合征;加速期;化疗;儿童
Chediak-Higashi综合征(以下称CHS)是一种较为罕见的常染色体隐性遗传性疾病,以部分性眼皮肤白化病、免疫缺陷、轻度的出血倾向及神经系统病变为主要表现,患者白细胞胞浆内见粗大包涵体颗粒具有临床诊断意义;患者一旦进入“加速期”,死亡率高,需要化疗。至1989年国外文献报道200余例[1],近15~20年共报道不到500例[2]。国内至今共报道50余例,文献多为个案或少量病例报道。由于发病率低,临床医师对本病认识还不足。此外,目前临床上对于“加速期”的诊断及给予化疗的时机并不明确。本文报道1例CHS并结合国内报道的病例资料,旨在总结CHS临床特点,重点探讨加速期的诊断及化疗时机。
病例资料,患儿女,4岁,2010年5月4日因“发热8天”入院。体温最高达41℃,无寒战、抽搐,无皮疹,发热时间不规律,热退精神尚可,输一般抗生素治疗(具体不详)无明显好转。3天前开始有单、双声咳嗽,不剧,痰少,无喘息、气促,无呼吸困难。患病后精神食纳可,无消瘦,大小便正常。
既往史:出生后约1~2月开始,渐出现皮肤颜色变化,出现颜面、颈部皮肤色素沉着,毛发色素沉着不良,颜面部分皮肤散在白斑。既往常因“感冒”就诊,多次查粒细胞减少,经一般抗感染治疗可治愈。生产史,喂养史,生长发育史正常。1+岁开始有畏光表现。家族史:患儿G5P3,人工流产2个,P1女性,皮肤病变与该患儿相似,自1-2岁开始反复出现皮肤化脓性感染,伴间断发热,予抗感染等治疗,反复不愈,至15岁病逝。P2女性,现17岁,上初中,无明显异常。父母非近亲结婚。
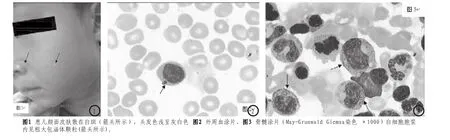
入院查体:T 37.4℃,P 104次/分,R 24次/分,Wt 15Kg,BP 80/50mmHg;神清,精神反应可;颜面、颈部、四肢皮肤见色素沉着及散在色素缺失斑(图1),毛发色素沉着不良(呈灰色);颈部可触及数枚黄豆大小不等淋巴结,质中活动度可,无红肿触痛;畏光,结膜无充血,眼球无震颤,瞳孔对光反射灵敏,唇无发绀,咽充血,扁桃体II°,见2个白色点状渗出;颈软,双肺呼吸音粗,未闻及干湿啰音;心音有力,律齐;腹软,肝右肋下平脐,质中缘锐,脾肋下3cm,质中,无压痛,肠鸣音正常;四肢活动自如,肌力、肌张力正常。神经系统查体:颅神经查体无明显异常,腱反射亢进,踝阵挛阳性,脑膜刺激征 阴性,巴氏征 阴性。
辅助检查:2010年5月2日血常规WBC3.5×10^9/ L,NEU0.35×10^9/L,RBC 3.78×10^9/L,Hb100g/ L,PLT81×10^9/L;CRP13mg/L。ESR 10mm/h。血涂片中性粒细胞0.09,淋巴细胞0.82,单核细胞0.08,异型淋巴细胞0.01,细胞形态大致正常。入院后查胸片示双肺纹理增多。腹部B超示肝脏、脾脏弥漫性增大。甘油三脂:1.82mmol/l,稍高。血清铁蛋白241.2ng/ml、凝血四项、血浆纤维蛋白原 2.305g/ l,基本正常。肝肾功、电解质、心肌酶、体液免疫、淋巴细胞分类计数基本正常。自身抗体 阴性。血清皮质醇13.58ug/dl;ACTH31.2pg/ml,铜蓝蛋白41.1mg/dl,正常范围。血EBV-DNA 6.33E+7,咽拭子EBV-DNA 5.29E+6阳性。肺炎支原体抗体阴性、咽拭子肺炎支原体DNA阴性。肥达试验阴性。结核杆菌抗体阴性。血培养阴性。骨髓涂片示 骨髓增生明显活跃。粒、红、巨系比例基本正常。粒系、淋系、单系细胞可见大小多少不一的包涵体,以粒系细胞为主(图2、图3)。全片可见少量组织细胞,部分吞噬白细胞。
治疗经过:入院后先给予哌拉西林他唑巴坦抗感染2天,热峰无下降。2010.05.06血常规WBC 3.7×10^9/L,NEU 0.58×10^9/L,Hb 99g/L,PLT 117×10^9/L,CRP 7.0mg/L。改为夫西地酸钠+头孢甲肟抗感染,2010.05.09加阿昔洛韦抗病毒,热峰有所下降。2010.05.10血常规WBC1.8×10^9/ L,NEU0.32×10^9/L,Hb96g/L,PLT65×10^9/L,CRP 30mg/L。2010.05.11停夫西地酸钠 改万古霉素+头孢甲肟+口服阿奇霉素抗感染,继续阿昔洛韦抗病毒,发热间隔有所延长。患儿于2010.05.14热退,2010.05.16血常规WBC 2.9×10^9/L,NEU 0.39×10^9/L,Hb 101g/L,PLT 144×10^9/L,CRP 6.0 mg/L,次日出院。
出院诊断:1、Chediak-Higashi综合征2、EB病毒感染3、急性支气管炎。
1 对象和方法
1.1 对象 以“Chediak-Higashi”或“先天性白细胞颗粒异常综合征”为关键词和主题词检索中国生物医学文献数据库(CBM)、维普、万方及CNKI(~2013年2月)全文数据库,收集病例报道,排除重复报道病例后纳入研究。
1.2 方法 收集每份文献中病例的相关临床资料。包括:性别、年龄、主诉、家族史、查体、检查、治疗、转归等,总结描述或统计分析。根据资料特点选择X2检验或t检验,使用SPSS 16.0统计软件,P<0.05差异有统计学意义。
2 结果
34篇文献纳入研究,其中2000年以前文献5篇,共计52例病例(包括本例)。
2.1 一般资料 男:女比例1.17:1(28:24)。诊断时年龄2.5(1~5)岁,最小42天,最大11岁。25%(8/32)家族史。25.8%(8/31)父母近亲结婚史。
2.2 临床表现 就诊主诉发热占92.3%(48/52);发热时间数天至反复1年不等,多有反复感染病史。1例42天患儿因体检发现。其次主诉或伴随主诉局部包块(入院后检查发现为肝/脾/淋巴结大)22.2%(8/36),皮肤、毛发颜色异常13.9%(5/36),咳嗽11.1%(4/36)。查体皮肤或毛发色素异常97.9%(46/47),皮肤多描述为棕黑色(色素沉着),散在白斑(色素脱失斑);毛发颜色多描述为灰白。眼睛表现畏光17.4%(4/23),眼球震颤39.1%(9/23),虹膜色素减淡69.6%(16/23)。其中1例眼及皮肤、毛发均无白化病表现。23.1%(12/52)就诊时有出血(皮肤瘀斑、瘀点或其它部位出血)症状。7.7%(4/52)神经系统异常,2例抽搐、双眼震颤,1例轻度脑萎缩,1例智力落后。
78.8 %(41/52)就诊时淋巴结大;86.5%(45/52)肝脏增大,平均(3.75±1.76)cm;86.5%(45/52)脾脏增大,(5.23±2.71)cm。30.9%(13/42)血常规三系(红细胞、粒细胞、血小板)减少,33.3%(14/42)两系减少,26.2%(11/42)一系减少,9.5%(4/42)正常。所有病例血涂片或骨髓检查可见白细胞胞浆内嗜酸性包涵体,为粗大颗粒,多描述为紫红色,其次粉红、桔红、灰褐色等。骨髓检查2例偶见噬血组织细胞,4例易见噬血细胞(噬血现象)。3例合并EB病毒感染。
2.3 治疗及转归 骨髓检查44例中,2例偶见噬血细胞,均未化疗,1例好转,1例无效。4例易见噬血细胞,其中1例VP(长春新碱+泼尼松)方案化疗,好转;1例未化疗,仅给予抗感染等治疗,热退,放弃治疗,2月后死亡;2例治疗不详,1例数月后死亡,1例好转。另有2例给予甲强龙、丙球治疗,好转。
29例患者中,化疗(化疗组,包括2例联合使用大剂量激素及丙球治疗病例)9例,9例好转(治疗好转率100%)。20例未化疗(未化疗组),仅给予一般抗感染等治疗,9例好转,11例无效或死亡(治疗好转率50%)。两组治疗好转率比较,X2=7.957,P=0.005,差异有统计学意义。
29例患者中,18例好转(好转组),11例无效/死亡患者(无效/死亡组)。如表1所示,无效/死亡组年龄较好转组小,脾脏较大,差异有统计学意义;无效/死亡组肝脏有增大趋势(P=0.097)。血常规两系降低的比例在两组间比较无统计学意义(表1)。
3 讨论
Chediak-Higashi综合征(CHS)是一种较为罕见的常染色体隐性遗传性疾病,又称为先天性白细胞颗粒异常综合征、异常白细胞包涵体综合征等。1943年Beguez Cesar等首次报道,1995年Sato认为Chediak和Higashi报告的资料相似,故命名。CHS与溶酶体转运调节因子基因(lysosomal trafficking regulator gene, LYST)突变有关。异常的LYST蛋白使囊泡转运调节异常,细胞内生成粗大溶酶体,异常溶酶体不能被转运到正常作用位点,从而引发各系统的临床症状。患者以部分性眼、皮肤白化病、免疫缺陷、轻度的出血倾向及神经系统病变为主要表现,白细胞胞浆内见粗大包涵体颗粒具有临床诊断意义。本病无特异治疗,骨髓移植可以改善免疫缺陷、出血症状,但不能阻止神经系统病变进展。由于反复感染、出血和“加速期”多器官病变,80%的患儿死亡,少数存活到成年[3]。本例既往反复感染、皮肤色素脱失及骨髓涂片白细胞胞浆内见包涵体颗粒,结合家族史,临床可确诊CHS。
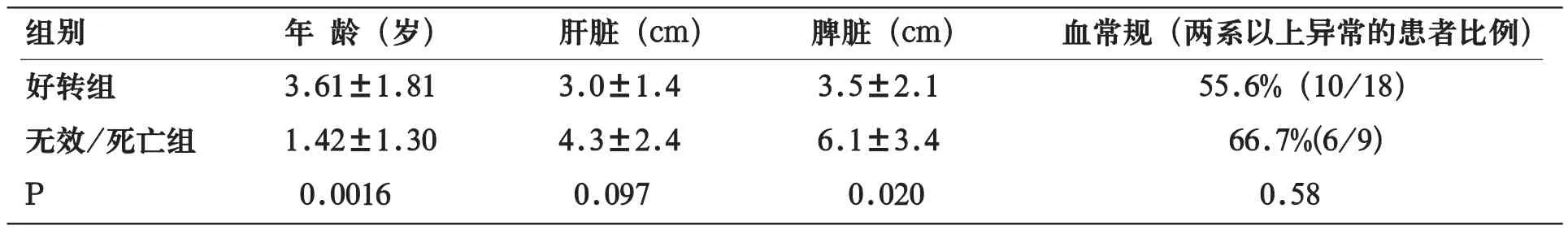
表1 好转组与无效/死亡组患者肝脏、脾脏大小及血常规检查比较
本研究显示,男:女患者比例为1.17:1。发病年龄可早至生后不久(如本例生后1~2月即开始皮肤改变),多为婴、幼儿时期发病。25%患者有家族史,25.8%患者父母近亲结婚。由于文献资料完整性参差不齐,未描述家族史及父母近亲结婚史的文献应多为无相关病史,因此实际家族史及父母近亲结婚史可能低于以上数据。资料显示,反复感染是患者最常见的表现及就诊原因,其次为皮肤色素异常及包块(肝/脾/淋巴结肿大)。查体皮肤多为暴露部分色素沉着,呈棕黑色,散在白斑(色素脱失斑)。眼睛畏光,眼球震颤,虹膜色素减退。出血症状较少报道,表现为皮肤瘀斑、瘀点。文献也较少提及神经系统症状及体征,可能由于神经系统症状呈进展性,多见于青少年、成人患者[4]。
部分性眼皮肤白化病,尤其是虹膜色素减少,既往曾作为特异性诊断指标。本组资料证实绝大部分患者均有突出的眼、皮肤白化病表现。但也有报道1例眼及皮肤均无白化病表现[5],该患者通过血细胞和骨髓涂片诊断。因此对于无明显白化病表现,但反复感染提示免疫缺陷的患者,仍需注意CHS。血涂片/骨髓涂片白细胞胞浆见粗大溶酶体颗粒是目前临床诊断CHS的指标,本组所有病例均通过血涂片/骨髓涂片诊断。近年发现细胞学改变程度与临床严重程度呈正比,融合的溶酶体颗粒越大,临床表现越重[6]。光学显微镜下头发色素减少有助于诊断,国内文献较少报道[7,8]。基因诊断CHS目前国内仅见重庆儿童医院临床免疫中心报道3例[7],该团队还探讨了流式细胞仪检测CD107α表达用于吞噬细胞功能缺陷病的诊断,并在3例CHS患者中得到验证[9]。
约85% CHS患者病情发展,进入“加速期”,表现为以多器官炎症为特征的噬血细胞性淋巴组织细胞增生症(HLH)[3]。HLH的指征包括[10]:1、发热 2、脾大 3、血细胞至少两系减少 4、肝功能异常表现:高甘油三酯血症和/或低纤维蛋白原血症 5、血清铁蛋白升高 6、血清可溶性IL-2受体升高 7、NK细胞活性降低 8、骨髓、脾脏、淋巴结或脑脊液中噬血现象(hemophagocytosis)。目前没有CHS“加速期”的诊断标准,国内文献也很少诊断“加速期”。本研究发现,治疗无效/死亡组(考虑其多为加速期病例)与治疗好转组相比,脾脏明显增大(P=0.02),肝脏有类似趋势(P=0.098),有助于临床诊断,而血常规两系降低比例在两组间无差异,考虑急性感染等因素可能使白细胞升高而影响该指标。但仍需注意到,本组资料显示绝大部分CHS患者反复发热,肝、脾、淋巴结肿大,血常规两系以上异常,提示以上指标对于判断加速期的意义有限。因此在参考HLH诊断标准时,其中脾肿大等指标可能需要探讨更具体的数据。由于病例资料不全,高甘油三酯血症、低纤维蛋白原血症以及血清铁蛋白升高等指标对判断加速期的价值有待进一步探讨。
CHS一般治疗为抗感染和对症。加速期是CHS患者死亡主要原因,需按家族性HLH化疗方案治疗[11]。本组资料表明化疗药物治疗组有效率显著高于仅给予一般抗感染治疗组,提示化疗有助于控制病情。CHS加速期可发生于任何年龄,甚至早至生后数月,临床表现为发热,肝、脾、淋巴结肿大,贫血、中性粒细胞或血小板减少[11]。但如上所述,本组资料表明大部分CHS患者就诊时有发热,肝、脾、淋巴结肿大,血常规两系以上异常,似乎为“加速期”表现,而报道骨髓检查发现噬血现象的比例并不高。本例CHS持续高热超过半月一般治疗效果欠佳、肝脾明显肿大,血象三系降低,骨髓可见部分组织细胞吞噬白细胞,尤其合并EB病毒感染,临床提示“加速期”的HLH。但肝功能基本正常、血清铁蛋白不高又不支持;且经抗感染及对症治疗最终病情得以控制也说明并没有进展到HLH。因此,这种情况给是否诊断CHS“加速期”以及临床决策是否给予化疗(包括丙球、激素、免疫抑制剂等)带来困难。CHS根据发病年龄可分为儿童型和青少年/成人型,前者起病早,感染症状更重,后者感染较轻而神经系统症状更明显[6]。本研究发现治疗无效/死亡组与治疗好转组相比,年龄明显较小,多为小婴儿,提示在儿童型患者中起病越早(指反复感染表现),病情越重,预后不良。因此,年龄可能作为CHS患者是否化疗的重要参考指标。对于1岁以内发病、进展,临床提示噬血细胞综合征表现的CHS患者,应积极考虑化疗。
[1] Introne W, Boissy R E, Gahl W A. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome[J]. Molecular genetics and metabolism, 1999, 68(2): 283-303.
[2] Kaplan J, De Domenico I, Ward D M V. Chediak-Higashi syndrome[J]. Current opinion in hematology, 2008, 15(1): 22-29.
[3] BLUME R S, WOLFF S M. The Chediak-Higashi syndrome: studies in four patients and a review of the literature[J]. Medicine, 1972, 51(4): 247-280.
[4] Tardieu M, Lacroix C, Neven B, et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrowtransplantation for Chediak-Higashi syndrome[J]. Blood, 2005, 106(1): 40-42.
[5] 苏绍东, 田开新. Chediak-Higashi 综合征一例报告[J]. 临床误诊误治, 2009, 2(3):39.
[6] Westbroek W, Adams D, Huizing M, et al. Cellular defects in Chediak-Higashi syndrome correlate with the molecular genotype and clinical phenotype[J]. Journal of Investigative Dermatology, 2007, 127(11): 2674-2677.
[7] 刘征, 蒋利萍, 刘玮, 等.4 例 Chediak-Higashi 综合征的临床特征和 LYST 基因分析[J]. 第三军医大学学报, 2011, 33(17): 1838-1842.
[8] 张新颖, 尹洪臣, 包静娴. Chediak-Higashi 综合征 2 例[J]. 中国当代儿科杂志, 2003, 5(4): 385-385.
[9] 王晶, 刘征, 蒋利萍, 等. CD107α 表达对 NK 细胞与细胞毒性T 细胞细胞毒功能缺陷性疾病筛查价值探讨[J]. 中华儿科杂志, 2012, 50(005): 386-391.
[10]Filipovich A H. Hemophagocytic lymphohistiocytosis and related disorders[J]. Current opinion in allergy and clinical immunology, 2006, 6(6): 410-415.
[11]Eapen M, DeLaat C A, Baker K S, et al. Hematopoietic cell transplantation for Chediak-Higashi syndrome[J]. Bone marrow transplantation, 2007, 39(7): 411-415.
Diagnosis and Treatment of Chediak-Higashi Syndrome: A Case Report and Literature Review
CHEN Jie-hua1, LI Chang-gang2, SHI Hong-song2,et al., 1 Department of Respiratory Disease; 2 Hematology, 4Clinical Laboratory, Shenzhen Children’s Hospital, 518026; 3Department of pediatric, Shenzhen Sixth People's Hospital, 518052
Objective To investigate the diagnosis and treatment of Chediak-Higashi syndrome(CHS) in accelerated phase. Methods Report a case and search the case reports of Chediak-Higashi syndrome in domestic literature, and collect the clinical datas for statistical analysis.Results 34 articles, a total of 52 cases were recruited. Male: female ratio of 1.17:1; Twenty five percent patients had family history; in 25.8% consanguineous marriage. 92.3% patients were hospitalized because of fever, many of them had repeated infection history, 97.9% had skin or hair pigmentation disorders; 78.8% had enlarged lymph nodes, 86.5% had splenomegaly and enlargement of liver. More than 64.2% patients had cytopenias (affected more than two lineages). The giant inclusions in the cytoplasmic of white cells in blood or bone marrow smear were found in all cases. The improvement rate in Chemotherapy group was higher than in Un-chemotherapy group (100% vs 50%, P=0.005). Invalid and death group compared with improvement group, age (years) (1.42 ± 1.30 vs 3.61 ± 1.81 P =0.0016), spleen (cm) (6.1 ± 3.4 vs 3.5 ± 2.1, P=0.02), liver (cm) (4.3 ± 2.4 vs 3.0 ± 1.4, P=0.097), cytopenias (affecting more than two of three lineages in the peripheral blood) (66.7% vs 55.6%, P=0.58).Conclusion Most of CHS patients showed recurrent fever, splenomegaly, enlargement of liver and lymph nodes, cytopenias (affected more than two lineages), which were not enough for diagnosis of a accelerated phase. The age of onset could be indication of chemotherapy, early onset of illness was associated with high mortality and chemotherapy helped to control the disease.
Chediak-Higashi Syndrome; Accelerated Phase ; Chemotherapy; Children
R179
A
2015-03-26
陈杰华,男,小儿呼吸专业,主治医师,主要研究方向为小儿呼吸与临床免疫。
刘四喜
DOI∶10.3969/j.issn.1009-3257.2015.02.007
猜你喜欢
娃娃乐园·综合智能(2022年9期)2022-08-16
健康体检与管理(2022年2期)2022-04-15
中国宝玉石(2021年5期)2021-11-18
中老年保健(2021年8期)2021-08-24
科学大众(2021年9期)2021-07-16
人人健康(2017年19期)2017-10-20
中国调味品(2017年2期)2017-03-20
现代食品(2016年24期)2016-04-28
中国继续医学教育(2015年3期)2016-01-06
转化医学电子杂志(2015年4期)2015-12-27
