
表达3-甾酮-△1-脱氢酶降解植物甾醇合成雄甾-1,4-二烯-3,17-二酮
2015-06-19 06:57:46张乐乐张显邵明龙陈榕榕饶志明李会许正宏
生物工程学报 2015年11期
张乐乐,张显,邵明龙,陈榕榕,饶志明,李会,许正宏
1 江南大学生物工程学院 工业生物技术教育部重点实验室,江苏 无锡 214122 2 江南大学药学院,江苏 无锡 214122
甾体类药物是仅次于抗生素的一大类药物,广泛应用于抗肿瘤、消炎、抗菌、抗病毒、抗真菌、抗雌激素、抗惊厥等领域[1]。全球每年对甾体类药物的需求量超过100万t,总价值超100亿美元[2]。甾体药物中间体的合成一般采用微生物转化法[3-4]。植物甾醇作为合成甾体激素类药物中间体的原料,具有廉价易得,微生物易于降解其侧链等特点[5-6]。微生物代谢植物甾醇的主要中间产物有雄甾-4-烯-3,17-二酮(4-androstene-3,17-dione, AD)、雄甾-1,4-二烯-3,17-二酮 (Androst-1,4-diene-3,17-dione,ADD)、睾酮、宝丹酮、9OH-AD等,其中AD和ADD市场需求量最高,因此利用廉价的底物植物甾醇高效合成AD和ADD具有巨大的商业价值[7]。
3-甾酮-△1-脱氢酶 (KSDD,EC 1.3.99.4)在甾体化合物代谢中具有重要的作用[8-11],它催化AD脱去 2个氢生成 ADD,同时也可催化9OH-AD合成9OH-ADD,9OH-ADD随后自发打开母核进而被进一步降解成 CO2和 H2O。基因敲除实验已经验证KSDD是甾醇代谢过程中关键酶,敲除 KSDD的编码基因将不能获得ADD[12-15]。目前,由于没有合适的工具——表达载体,利用分子生物学方法对分枝杆菌进行改造生产ADD比较困难[16]。虽然已有对KSDD同源加强表达的研究,但并未给出具体的酶活数据及蛋白表达情况[15,17]。分枝杆菌Mycobacterium neoaurum能够降解植物甾醇合成激素类药物中间体[18-21]。M. neoaurumJC-12是本研究室从土壤中筛选到的菌株,具有降解植物甾醇合成AD和ADD的能力。实验室前期克隆表达出M. neoaurumJC-12的ksdd基因,并对KSDD的转化能力进行了研究[22]。在分枝杆菌中加强表达KSDD是促进ADD高效生产的一个重要途径。然而,分枝杆菌表达系统还不完善,缺乏高效表达载体,这已成为目前分枝杆菌降解植物甾醇合成药物中间体研究中的瓶颈。tac启动子是trp启动子和lacUV5的拼接杂合启动子,在大肠杆菌中具有很高的转录水平,用于钝齿棒杆菌、黄色短杆菌和谷氨酸棒杆菌等阳性菌表达基因,也有较高的表达效率[23]。
所以,本研究构建了具有tac启动子的分枝杆菌表达载体,并通过表达绿色荧光蛋白 GFP验证其在M. neoaurumJC-12中的高效表达,为后续在分枝杆菌中高效表达KSDD奠定基础。
1 材料与方法
1.1 菌株与质粒
M. neoaurumJC-12由本实验筛选并保存,Escherichia coliJM109及用于提供绿色荧光蛋白报告基因的质粒pCAMBIA1302和pETac由本实验保存,分枝杆菌表达载体pMF41由上海公共卫生中心范小勇博士惠赠。
1.2 培养方法
1.2.1 大肠杆菌的培养
LB培养基 (g/L):蛋白胨10,酵母提取物5,NaCl 10,如需固体培养基,加入2%琼脂。旋转式摇床160 r/min,37 ℃,转化子筛选条件:加入卡那霉素终浓度为50 μg/mL。
1.2.2 分枝杆菌的培养
活化培养基 (g/L):葡萄糖 5,蛋白胨 5,牛肉膏5,甘油15,NaCl 15,pH 7.0。
摇瓶发酵培养基 (g/L):葡萄糖 20,蛋白胨 10, K2HPO4·3H2O 3, MnCl20.000 5 ,MgSO4·7H2O 0.2,植物甾醇 10,环糊精 30,pH 7.0 (250 mL三角瓶,装液量50 mL)。感受态制备培养基:LB + 0.2% Tween 80。旋转式摇床转速为160 r/min,30 ℃,转化子筛选条件:加入卡那霉素终浓度为30 μg/mL。
发酵罐培养基 (g/L):植物甾醇20,环糊精60,其余同摇瓶发酵培养基 (5 L发酵罐,装液量2 L)。转速400 r/min,30 ℃,通气量1 vvm,发酵过程中流加葡萄糖 (母液浓度为800 g/L)。
1.3 主要试剂与仪器
基因组提取试剂盒、质粒提取试剂盒、胶回收试剂盒、抗生素、PCR引物、2,6-二氯酚靛酚 (DCPIP)及吩嗪硫酸甲酯 (PMS) 等购自上海生工生物工程有限公司;Taq酶、限制性内切酶、T4 DNA连接酶等购自TaKaRa公司;植物甾醇 (大豆甾醇≥95%) 购自礼来生物技术 (湖州) 有限公司;ADD及AD标准样品购自Sigma公司。PCR仪为BIO-RAD C1000;电转化仪为BIO-RAD pulse controller (Gene Pulser);荧光显微镜为Nikon ECLIPSE 50。
1.4 表达载体pMTac的构建
根据tac启动子的序列设计引物 (表 1):tac-F和tac-R。以pETac载体为模板,以tac-F和tac-R为引物,进行PCR扩增。条件为:95 ℃变性 3 min;95 ℃变性 30 s,60 ℃复性 30 s,72 ℃延伸30 s,30个循环;72 ℃保温5 min。PCR产物和质粒pMF41经XbaⅠ和BamHⅠ双酶切,T4 DNA连接酶16 ℃连接纯化后的产物,转化大肠杆菌JM109,阳性转化子酶切电泳验证。
1.5 tac启动子在分枝杆菌中的表达
1.5.1 绿色荧光蛋白表达菌株的构建
根据GenBank中登记的绿色荧光蛋白基因gfp设计引物 (表1):gfp-F1、gfp-F2和gfp-R。以 pCAMBIA1302为模板,分别以gfp-F1和gfp-R、gfp-F2和gfp-R为上下游引物,PCR扩增片段经双酶切后分别连接到经同样双酶切的pMF41和pMTac上,得重组载体pMF41-gfp和pMTac-gfp。采用电击转化法将空载体及重组载体电转到M. neoaurumJC-12中,用终浓度为30 μg/mL的固体培养筛选阳性转化子,并提质粒酶切验证。
1.5.2 GFP的荧光检测
重组菌pMF41-gfp培养至OD600为0.8左右时,加入终浓度为0.02%乙酰胺诱导,继续培养24 h。重组菌pMTac-gfp不需诱导。取重组菌发酵液,离心收集细胞,50 mmol/L Tris-HCl (pH 7.0)缓冲液洗涤 3次,重悬,稀释至适当浓度取少许于载玻片上,盖上盖玻片,在Nikon ECLIPSE 50荧光显微镜下观察。以原始菌M. neoaurumJC-12作为对照。
1.6 3-甾酮-△1-脱氢酶加强表达
1.6.1 3-甾酮-△1-脱氢酶加强表达菌株的构建
根据分枝杆菌 JC-12的 3-甾酮-△1-脱氢酶编码基因ksdd设计引物 (表1):ksdd-F1、ksdd-F2和ksdd-R。以分枝杆菌JC-12基因组DNA为模板,同上得到重组表达载体 pMF41-ksdd和pMTac-ksdd,并电转化至M. neoaurumJC-12中。
1.6.2 KSDD表达及酶活测定
重组菌pMF41-ksdd培养至OD600为0.8左右时,加入终浓度为0.02%乙酰胺诱导,继续培养24 h。重组菌pMTac-ksdd不需诱导。取重组菌发酵液,离心收集细胞,50 mmol/L Tris-HCl(pH 7.0) 缓冲液洗涤 3次,重悬,超声波破碎细胞,8 000 r/min离心30 min后得破碎细胞上清液。12% SDS-PAGE分析蛋白表达情况。

表1 本研究所用的引物Table 1 Primers used in this research
采用 PMS-DCPIP法测定 KSDD酶活。KSDD脱去底物AD的两个氢生成ADD,蛋白自身的FAD接受脱去的2 H+和2 e–成FADH2,然后FADH2将DCPIP还原,DCPIP在600 nm处有吸收峰。3 mL反应混合物由100 μL粗酶液、50 mmol/L Tris-HCl (pH 7.0)、40 μmol/L 2,6-二氯酚靛酚 (DCPIP)、1.5 mmol/L吩嗪硫甲酯(PMS)、500 μmol/L AD (溶于 2%的甲醇) 所组成,检测600 nm处的吸光值变化。酶活单位定义:将在1 min内还原1 μmol DCPIP所需的酶量定义为一个酶活单位 (U)。
1.7 重组分枝杆菌的发酵实验
1.7.1 摇瓶发酵条件
摇瓶发酵条件为:250 mL三角瓶,50 mL装液量,10%接种量,30 ℃,160 r/min。重组菌 pMF41-ksdd接种后培养 5 h添加终浓度为0.02%的乙酰胺。从发酵 48 h开始取样,每隔24 h取一次,共取样6次,即发酵周期为168 h。
1.7.2 5 L发酵罐发酵条件
将活化好的种子接种至2 L发酵培养基中,10%接种量,30 ℃,400 r/min,pH 维持在7.0−7.5。每隔12 h取一次样,测定葡萄糖浓度、菌体量[18]及AD和ADD的产量。
1.7.3 发酵产物的检测
分枝杆菌 JC-12降解植物甾醇的主要产物是AD和ADD。AD和ADD在254 nm处均有吸收峰,因此可以采用高效液相色谱法 (HPLC)对产物进行定量的分析。取1 mL发酵液,加入5 mL乙酸乙酯萃取,漩涡振荡充分混匀,0.22 μm尼龙滤膜过滤。HPLC条件:色谱柱:Dimosoil C18(250 mm×4.6 mm,5 μL);流动相:70%甲醇(V甲醇/V水=73)∶;检测器与检测波长:UV 检测器,254 nm;进样量:10 μL;柱温:30 ℃;流速:1.0 mL/min。
2 结果与分析
2.1 表达载体pMTac的构建
PCR所得大小约250 bp左右的tac启动子片段经BamHⅠ和XbaⅠ双酶切后,连接同样双酶切的载体 pMF41,得到新载体 pMTac。构建过程及酶切验证见图1。
2.2 tac启动子在分枝杆菌中强度的分析
2.2.1 绿色荧光蛋白表达菌株的构建
PCR所得的大小为760 bp左右的gfp基因片段经BamHⅠ和EcoRⅠ双酶切后,分别连接到相应的经BamHⅠ和EcoRⅠ双酶切的pMF41和 pMTac,获得重组载体 pMF41-gfp和pMTac-gfp。酶切验证见图2。通过电转化法将重组载体转化至M. neoaurumJC-12,获得重组菌。
2.2.2 GFP的荧光检测
取表达后的重组菌,缓冲液洗涤 3次,涂在玻璃载玻片上,荧光显微镜下观察,结果见图3。荧光照片的结果显示,原始菌M. neoaurumJC-12无荧光;M. neoaurumJC-12/pMF41-gfp和M. neoaurumJC-12/pMTac-gfp都有荧光亮度,但是M. neoaurumJC-12/pMTac-gfp的亮度明显高于M. neoaurumJC-12/pMF41-gfp,说明tac启动子可以用于分枝杆菌中表达基因,而且强度高于pACE。重组菌和对照菌的诱导表达条件及荧光显微镜观察条件均相同。
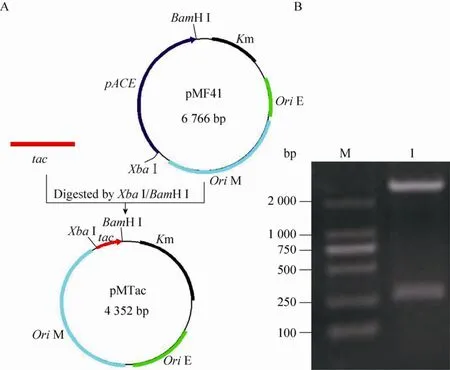
图1 新型载体pMTac的构建 (A) 及酶切验证 (B)Fig. 1 Construction (A) and identification (B) of the plasmid pMTac. M: DL2 000 DNA marker; 1: pMTac digested with BamH I and Xba I.
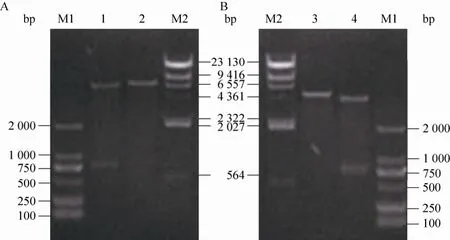
图2 重组质粒pMF41-gfp (A) 和pMTac-gfp (B) 的酶切验证Fig. 2 Identification of recombinant plasmid pMF41-gfp (A) and pMTac-gfp (B). M1: DL2 000 DNA marker; 1:pMF41-gfp digested with BamH I and EcoR I; 2: pMF41-gfp digested with BamH I; M2: λ-Hind Ⅲ DNA marker; 3:pMTac-gfp digested with BamH I; 4:pMTac-gfp digested with BamH I and EcoR I.
2.3 3-甾酮-△1-脱氢酶加强表达
2.3.1 3-甾酮-△1-脱氢酶加强表达菌株的构建
以提取的M. neoaurumJC-12基因组DNA为模板PCR得到ksdd基因片段,同上述方法获得重组菌M. neoaurumJC-12/pMF41-ksdd和M. neoaurumJC-12/pMTac-ksdd。酶切验证见图4。
2.3.2 KSDD的表达及酶活测定
重组菌M. neoaurumJC-12/pMF41-ksdd经乙酰胺诱导ksdd表达,M. neoaurumJC-12/pMTac-ksdd不需诱导,处理后获得破细胞上清液。SDS-PAGE验证KSDD表达情况(图5)。结果表明M. neoaurumJC-12/pMTac-ksdd的破细胞上清液与M. neoaurumJC-12相比在60 kDa附近有一条蛋白条带加粗,而且与这条蛋白条带与KSDD蛋白大小相同。M. neoaurumJC-12/pMF41-ksdd破细胞上清液与M. neoaurumJC-12相比没有明显加粗的条带。

图3 分枝杆菌JC-12细胞中绿色荧光蛋白的检测Fig. 3 Observation of GFP in M. neoaurum JC-12 and the recombinants. (A) M. neoaurum JC-12. (B) M. neoaurum JC-12/pMF41-ksdd. (C) M. neoaurum JC-12/pMTac-ksdd.
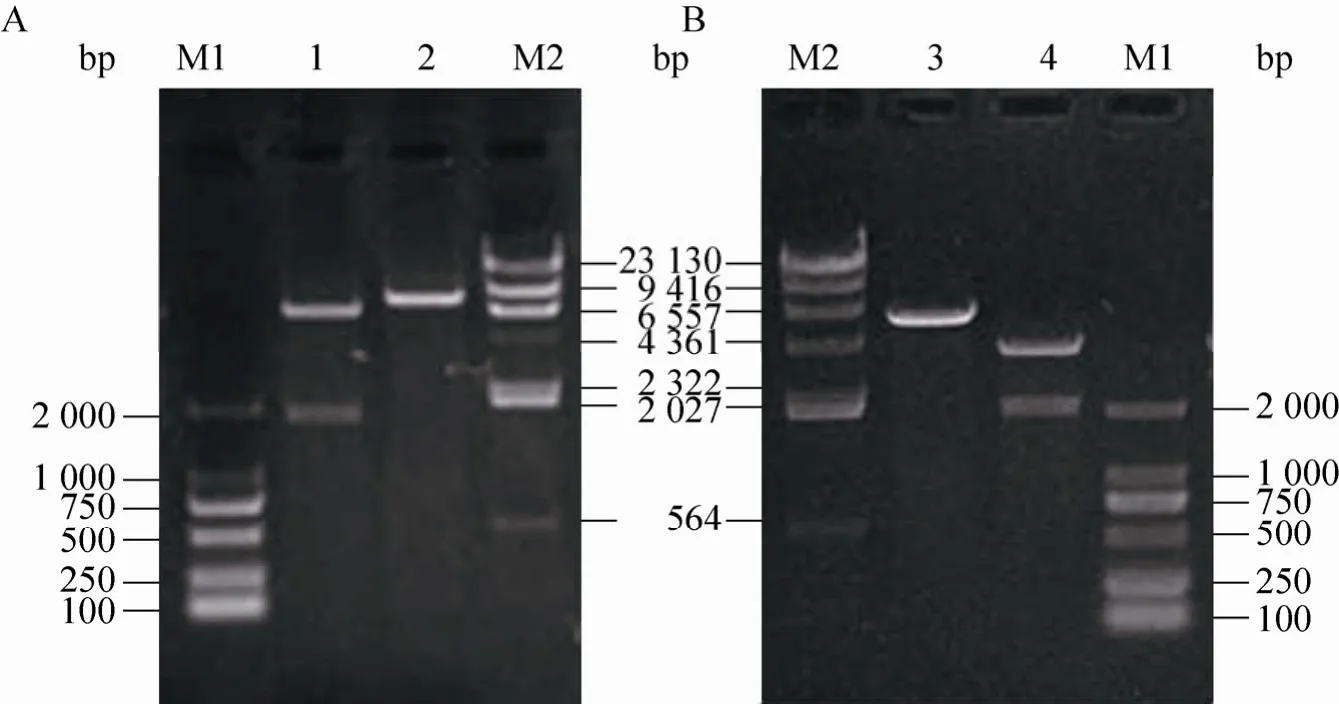
图4 重组质粒pMF41-ksdd (A) 和pMTac-ksdd (B) 的酶切验证Fig. 4 Identification of recombinant plasmid pMF41-ksdd (A) and pMTac-ksdd (B). M1: DL2 000 DNA marker; 1:pMF41-ksdd digested with BamH I and EcoR I; 2: pMF41-ksdd digested with BamH I; M2: λ-Hind Ⅲ DNA marker;3: pMTac-ksdd digested with BamH I; 4: pMTac-ksdd digested with BamH I and EcoR I.
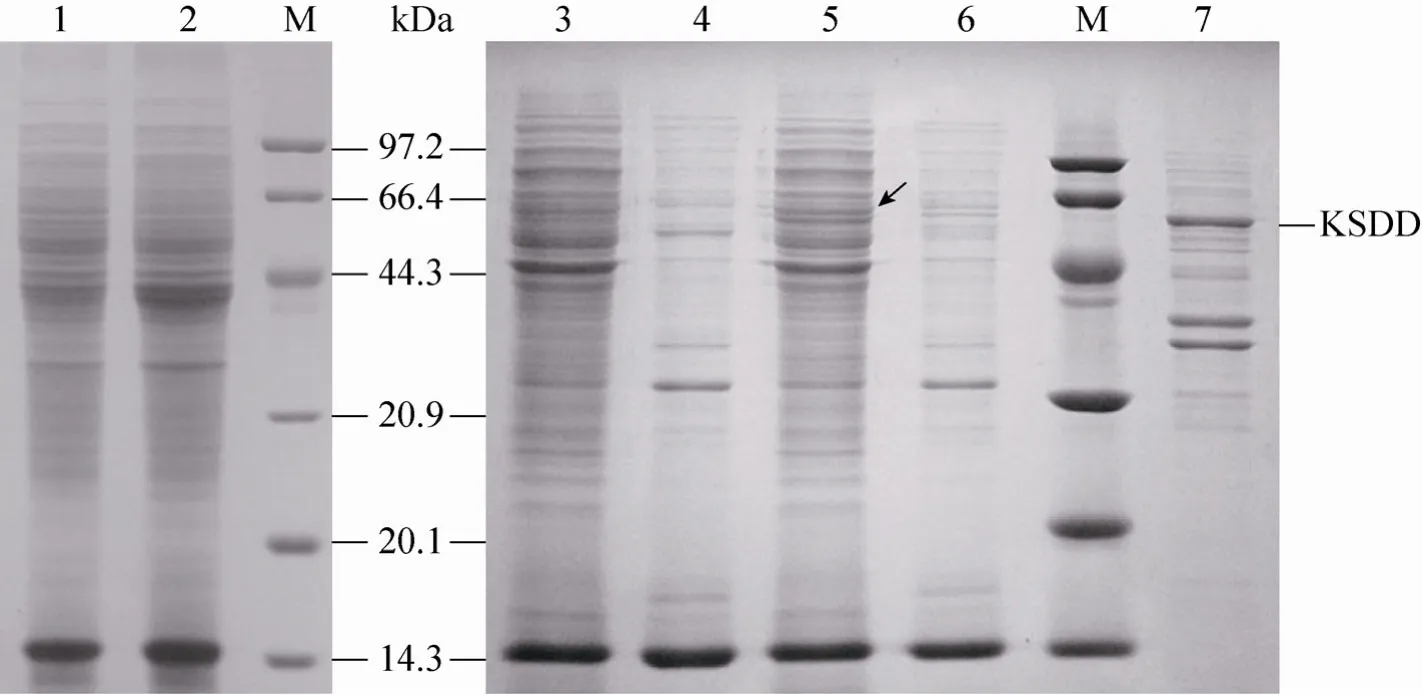
图5 重组菌蛋白的SDS-PAGE分析Fig. 5 SDS-PAGE analysis of the expressed protein. 1: the supernatant fraction of M. neoaurum JC-12/pMF41-ksdd;2, 3: the supernatant fraction of M. neoaurum JC-12; 4: the sediment fraction of M. neoaurum JC-12; 5: the supernatant fraction of M. neoaurum JC-12/pMTac-ksdd; 6: the sediment fraction of M. neoaurum JC-12/pMTac-ksdd;7: the sediment fraction of E. coli JM109/pMTac-ksdd; M: protein marker.
表达后的重组菌,超声波破碎细胞,离心得粗酶液,参照 1.6.2的方法测定粗酶液中KSDD的活性,结果见表2。重组菌M. neoaurumJC-12/pMTac-ksdd破细胞上清液中KSDD的活性为 (2.41±0.05) U/mg,比原始菌提高了6.53倍,而且较重组菌M. neoaurumJC-12/pMF41-ksdd提高了4.36倍,其中酶活单位U/mg的mg是指破细胞上清液即粗酶液中的蛋白含量。
酶活测定结果与重组菌的GFP荧光检测结果相一致,再次证明tac启动子在分枝杆菌中有作用且强度高于pACE,定量验证了tac启动子在分枝杆菌中强度。
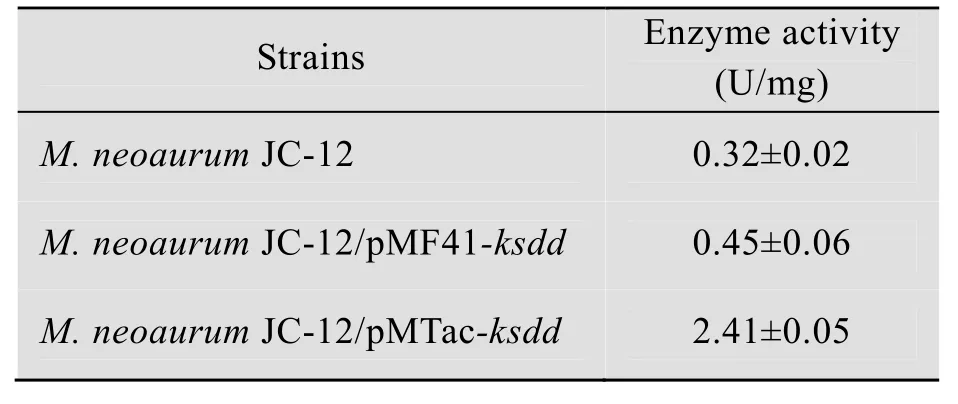
表2 重组菌株KSDD酶活的测定Table 2 The KSDD activities of recombinant cells
2.4 发酵结果
2.4.1 摇瓶发酵
以10 g/L植物甾醇为底物,环糊精为底物助溶剂进行发酵。菌株活化48 h后以10%的接种量接种到发酵培养基中,每组 3个平行,结果取平均值。
发酵结果如图 6,原始菌、重组菌M. neoaurumJC-12/pMF41- ksdd和M. neoaurumJC-12/pMTac-ksdd发酵7 d的ADD产量分别为4.86 g/L、5.27 g/L 和 5.94 g/L。重组菌M. neoaurumJC-12/pMTac-ksddADD的产量比原始菌提高了 22.2%,比M. neoaurumJC-12/pMF41-ksdd提高了12.7%;同时,AD的产量由原始菌的 0.92 g/L降低到M. neoaurumJC-12/pMTac-ksdd的0.17 g/L,重组菌AD降低了81.5%。发酵过程中ADD与AD的摩尔比值也产生了变化。整个发酵过程中重组菌M. neoaurumJC-12/pMTac-ksdd的ADD/AD值都高于原始菌,168 h时M. neoaurumJC-12/pMTac-ksdd的 ADD/AD 值为 34.9∶1,而原始菌的的ADD/AD值为5.3∶1。这是由于重组菌中加强表达了KSDD,它催化AD进一步转化成ADD。
从以上数据可以看出,加强表达KSDD不仅可以提高ADD产量,降低AD的量,还可以缩短发酵周期至少48 h。
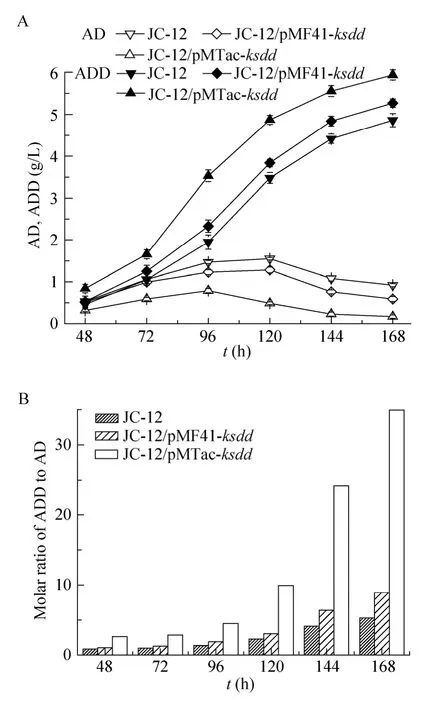
图6 摇瓶发酵结果Fig. 6 Datas of flask fermentation. (A) Production of AD and ADD. (B) The molar ratio of ADD to AD.
2.4.2 5 L发酵罐发酵结果
以20 g/L植物甾醇为底物,环糊精为底物助溶剂进行发酵。菌株活化48 h后以10%的接种量接种到发酵罐中。发酵72 h开始流加葡萄糖,维持葡萄糖浓度在4−5 g/L左右 (图7)。
发酵 168 h后重组菌M. neoaurumJC-12/pMTac-ksdd的ADD产量达到10.28 g/L,比原始菌提高了25.1%,AD的产量为0.24 g/L,比原始菌降低了80.6%。
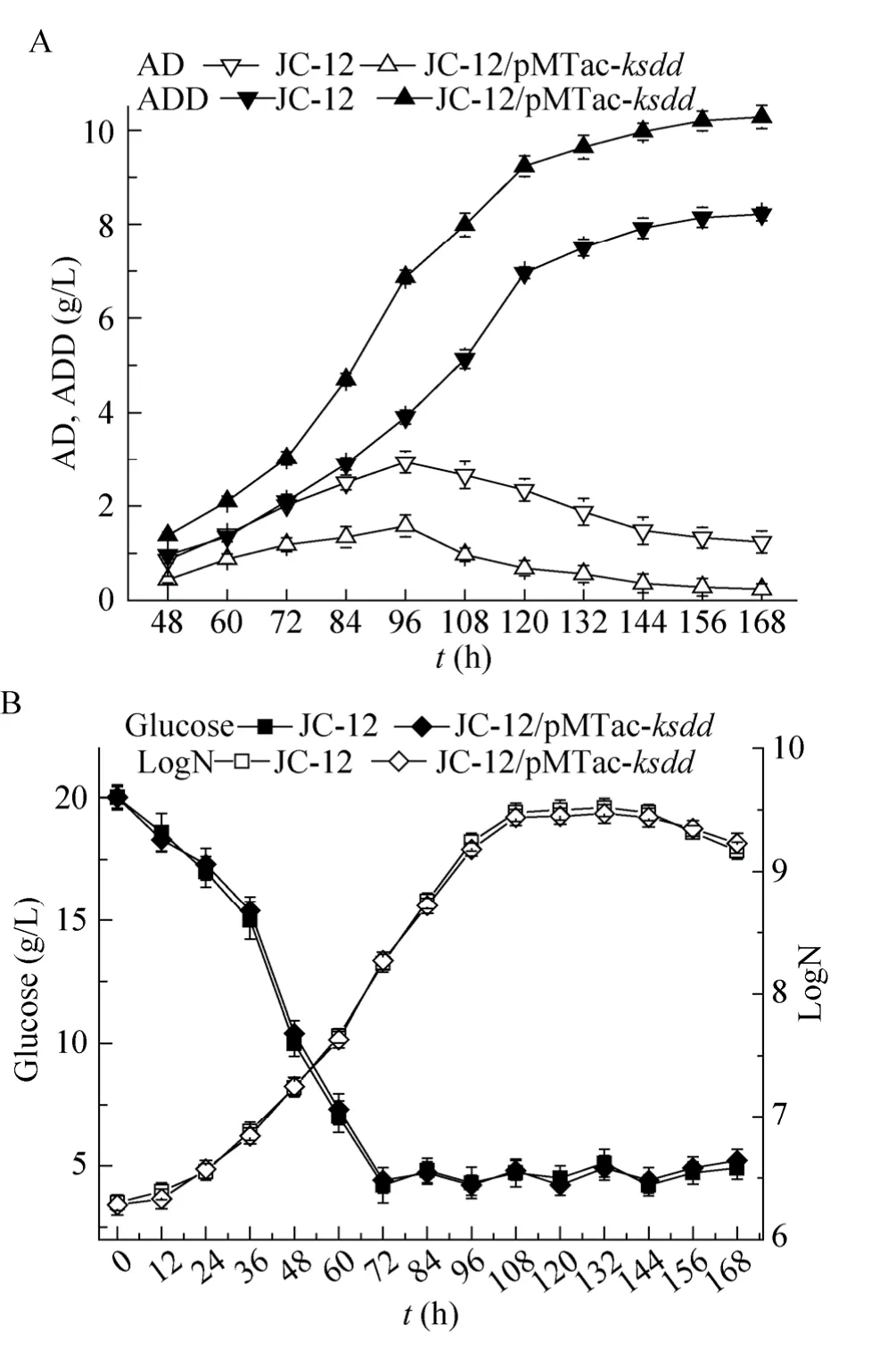
图7 5 L发酵罐发酵结果Fig. 7 Datas of 5 L fermenter fermentation. (A)Production of AD and ADD. (B) Concentration of glucose and strain growth, N is the number of CFU per milliliter of culture broth.
3 讨论
分枝杆菌可诱导表达载体pMF41由范小勇博士构建[24],用于在快速生长型分枝杆菌M.smegmatis中高水平表达分枝杆菌来源基因,但它在慢速生长型分枝杆菌中不能高水平表达蛋白,这可能是由于pMF41的启动子pACE来源于快速生长的M. smegmatis,且结构比较复杂。本研究室也通过实验证明pMF41不适用于慢速生长型分枝杆菌M. neoaurumJC-12表达蛋白,因此构建了表达载体 pMTac。本研究比较了耻垢分枝杆菌乙酰胺酶基因启动子pACE和广宿主的tac启动子对 3-甾酮-△1-脱氢酶基因ksdd在M. neoaurumJC-12中表达的影响,GFP荧光检测和KSDD酶活测定结果表明tac启动子在 JC-12中能够启动基因的表达且强度高于pACE。由于载体pMTac上没有lacI基因,所以pMTac在分枝杆菌中实施组成型表达,避免了诱导物IPTG对细胞的毒性,这样更有利于工程菌大规模工业化发酵。
2007年 Molchanova等报道M. neoaurumVKPM Ac-1656以10 g/L胆固醇为底物发酵得到5.5 g/L ADD,但是在相同条件下这株菌对植物甾醇的利用非常缓慢,发酵 5 d都未检测到ADD[18]。2010年Wei等报道了一株新金色分枝杆菌NwIB-01,以15 g/L植物甾醇为底物,发酵罐发酵96 h获得4.23 g/L ADD和1.76 g/L AD[21],并且随后通过加强表达关键酶 3-甾酮-△1-脱氢酶使ADD的产量提高至4.96 g/L[15]。2011年 Andryushina等通过添加助溶剂甲基-β-环糊精使M. neoaurumAc 1634转化30 g/L大豆甾醇合成AD达15.2 g/L,但是没有ADD的积累[25]。张小燕从土壤中筛选出一株可以植物甾醇为唯一碳源的新金色分枝杆菌M. neoaurumZJUVN (AD与ADD的产量比约为10∶1),使用 N+离子注入技术进行诱变得到一株单产 AD的菌株M. neoaurumZJUVN-08,随后对培养基及转化条件进行了优化,最终以8.89 g/L植物甾醇为底物转化得5.96 g/L AD[26-27]。
本研究室已研究KSDD的异源表达及转化性能,它是甾醇代谢中的关键酶,催化 AD合成ADD。一直以来,由于没有合适的表达载体,无法对M. neoaurumJC-12进行分子改造。而且产物AD和ADD的分离也是工业生产中的一大难题。新型表达载体pMTac的构建打破了这一束缚,不仅实现了基因的加强表达,使产物ADD的产量提高,AD的产量降低,并且缩短了发酵周期。
[1]Donova M, Egorova O. Microbial steroid transformations: current state and prospects. Appl Microbiol Biotechnol, 2012, 94(6): 1423−1447.
[2]Bureik M, Bernhardt R. Modern Biooxidation:Enzymes, Reactions and Applications. Weinheim:Wiley-VCH Verlag GmbH & Co. KGaA, 2007:155−176.
[3]Tong W, Dong X. Microbial biotransformation:recent developments on steroid drugs. Recent Pat Biotechnol, 2009, 3(2): 141−153.
[4]Liang JJ, Wang WJ. Progress of biotransforming phytosterols into androstadienedione and androstenedione by microorganisms. Hubei Agric Sci, 2012, 51(7): 1309−1312 (in Chinese).
梁建军, 汪文俊. 微生物生物转化甾体化合物生产雄烯二酮研究进展. 湖北农业科学, 2012,51(7): 1309−1312.
[5]Fernandes P, Cabral JMS. Phytosterols:applications and recovery methods. Bioresour Technol, 2007, 98(12): 2335−2350.
[6]Zhang YQ, Wang DQ. Advances in microbial transformation of phytosterol into steroid medicine intermediates. Microbiol China, 2006, 33(2):142−146 (in Chinese).
张裕卿, 王东青. 植物甾醇微生物转化制备甾体药物中间体的研究进展. 微生物学通报, 2006,33(2): 142−146.
[7]Malaviya A, Gomes J. Androstenedione production by biotransformation of phytosterols. Bioresour Technol, 2008, 99(15): 6725−6737.
[8]Itagaki E, Wakabayashi T, Hatta T. Purification and characterization of 3-ketosteroid-Δ1-dehydrogenase fromNocardia corallina. Prot Struct Mol Enzymol,1990, 1038(1): 60−67.
[9]Morii S, Fujii C, Miyoshi T, et al.3-Ketosteroid-Δ1-dehydrogenase ofRhodococcus rhodochrous: sequencing of the genomic DNA and hyperexpression, purification, and characterization of the recombinant enzyme. J Biochem, 1998,124(5): 1026−1032.
[10]Van der Geize R, Hessels GI, Dijkhuizen L.Molecular and functional characterization of thekstD2gene ofRhodococcus erythropolisSQ1 encoding a second 3-ketosteroid δ¹-dehydrogenase isoenzyme. Microbiol, 2002, 148(10): 3285−3292.
[11]Yin-Ru C, Ismail W, Gallien S, et al.Cholest-4-en-3-one-Δ1-dehydrogenase, a flavoprotein catalyzing the second step in anoxic cholesterol metabolism. ApplEnviron Microbiol, 2008, 74(1):107−113.
[12]Anna B, Śliwiń ski T, Rumijowska-Galewicz A, et al. Identification and targeted disruption of the gene encoding the main 3-ketosteroid dehydrogenase inMycobacterium smegmatis. Microbiology, 2005,151(7): 2393−2402.
[13]Van der Geize R, Hessels GI. Targeted disruption of the kstD gene encoding a 3-ketosteroid delta1-dehydrogenase isoenzyme ofRhodococcus erythropolisstrain SQ1. Appl Environ Microbiol,2000, 66(5): 2029−2036.
[14]Van der Geize R, Hessels GI, Van Gerwen R, et al.Unmarked gene deletion mutagenesis ofkstD,encoding 3-ketosteroid Δ1-dehydrogenase, inRhodococcus erythropolisSQ1 usingsacBas counter-selectable marker. FEMS Microbiol Lett,2001, 205(2): 197−202.
[15]Wei W, Wang FQ, Fan SY, et al. Inactivation and augmentation of the primary 3-ketosteroid-Δ1-dehydrogenase inMycobacterium neoaurumNwIB-01: biotransformation of soybean phytosterols to 4-androstene-3,17-dione or 1,4-androstadiene-3,17-dione. Appl Environ Microbiol, 2010, 76(13): 4578−4582.
[16]Mulder MA, Zappe H, Steyn LM.Mycobacterialpromoters. Tuber Lung Dis, 1997, 78(5): 211−223.
[17]Wei W, Fan SY, Wang FQ, et al. Accumulation of androstadiene-dione by overexpression of heterologous 3-ketosteroid Δ1-dehydrogenase inMycobacterium neoaurumNwIB-01. World J Microbiol Biotechnol, 2014, 30(7): 1947−1954.
[18]Molchanova M, Andryushina V, Savinova T, et al.Preparation of androsta-1,4-diene-3,17-dione from sterols usingMycobacterium neoaurumVKPM Ac-1656 strain. Russ J Bioorg Chem, 2007, 33(3):354−358.
[19]Rodina N, Molchanova M, Voishvillo N, et al.Conversion of phytosterols into androstenedione byMycobacteriumneoaurum. Appl Biochem Microbiol, 2008, 44(1): 48−54.
[20]Voishvillo NE, Andryushina VA, Savinova TS, et al. Identification of a new steroid-transforming strain ofMycobacteriaasMycobacterium neoaurum. Appl Biochem Microbiol, 2003, 39(2):152−157.
[21]Wei W, Fan S, Wang F, et al. A new steroid-transforming strain ofMycobacterium neoaurumand cloning of 3-ketosteroid 9α-hydroxylase in NwIB-01. Appl Biochem Biotechnol, 2010, 162(5): 1446−1456.
[22]Zhang W, Shao M, Rao Z, et al. Bioconversion of 4-androstene-3,17-dione to androst-1,4-diene-3,17-dione by recombinantBacillus subtilisexpressingksddgene encoding 3-ketosteroid-Δ1-dehydrogenase fromMycobacterium neoaurumJC-12. J Steroid Biochem Mol Biol, 2013, 135(1): 36−42.
[23]Xu DQ. Construction of vector systems forBrevibacterium flavumand its L-valine production metabolic engineering[D]. Wuxi: Jiangnan University, 2010 (in Chinese).
徐大庆. 黄色短杆菌载体系统的构建及其产L-缬氨酸代谢工程育种的初步研究[D]. 无锡: 江南大学, 2010.
[24]Fan XY. Establishment of the novel expression systems inMycobacteriaand their application for recombinant BCG[D]. Shanghai: Fudan University,2008 (in Chinese).
范小勇. 分支杆菌新型表达系统的建立及其在基因重组卡介苗研究中的应用[D]. 上海: 复旦大学, 2008.
[25]Andryushina VA, Rodina NV, Stytsenko TS, et al.Conversion of soybean sterols into 3,17-diketosteroids using actinobacteriaMycobacterium neoaurum,Pimelobacter simplex,andRhodococcus erythropolis. Appl Biochem Microbiol, 2011, 47(3): 270−273.
[26]Zhang XY. Studies on biotransformation from phytosterol to androstenedione byMycobacterium neoaurumZJUVN-08[D]. Hangzhou: Zhejiang University, 2013 (in Chinese).
张小燕. 新金分枝杆菌 ZJUVN-08转化植物甾醇合成雄甾烯二酮的研究[D]. 杭州: 浙江大学,2013.
[27]Zhang XY, Peng Y, Su ZR, et al. Optimization of biotransformation from phytosterol to androstenedione by a mutantMycobacterium neoaurumZJUVN-08. J Zhejiang Univ: Sci B(Biomed Biotechnol), 2013, 14(2): 132−143.
猜你喜欢
食品安全导刊(2022年32期)2022-12-07 05:36:08
华人时刊(2022年9期)2022-09-06 01:02:44
华人时刊(2020年15期)2020-12-14 08:10:36
心肺血管病杂志(2018年11期)2018-12-18 01:51:40
中国医药生物技术(2015年4期)2015-12-26 08:26:36
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
河南医学研究(2014年3期)2014-02-27 14:51:59
中国粮油学报(2014年8期)2014-02-06 01:34:23
中国火炬(2013年11期)2013-07-25 09:50:19
食品科学(2013年23期)2013-03-11 18:30:10