
双肾多发结石、高草酸尿症、肾功能不全
2015-06-09 06:47:20周玉超刘志红
肾脏病与透析肾移植杂志 2015年4期
周 岩 周玉超 刘志红 程 震
·临床集锦·
双肾多发结石、高草酸尿症、肾功能不全
周 岩 周玉超 刘志红 程 震
12岁男性,因“多发肾结石6年,血清肌酐升高8月,加重1月余”入院。患者临床表现双肾多发结石,缓慢进展至慢性肾功能不全;结石成分分析为“一水草酸钙”,其父亲患有肾结石。结合临床、实验室检查、GRHPR基因分析诊断为原发性高草酸尿症2型,高草酸尿症导致肾损害。
高草酸尿症 肾结石 基因诊断
病例摘要
12岁男性,因“多发肾结石6年余,血清肌酐(SCr)升高8月,加重1月余” 于2014-10-16入院。
现病史 患者2008年5月出现肋弓下缘外翻及肋骨串珠样改变,外院诊断“佝偻病”,B超示“双肾结石”,未予治疗。2008年10月查尿蛋白、隐血均为阴性,尿白细胞>500/HP,尿pH 6.5,比重1.01,尿钙0.7 mmol/L,尿磷5.77 mmol/L,SCr 61 μmol/L,血红蛋白(Hb)130 g/L,甲状旁腺激素(iPTH)3.2 pmol/L,双肾B超示“双肾椎体呈增强团块状回声,伴后声影”,静脉肾盂造影示“双肾区多发类圆形高密度影”。予留置双J管后多次行体外震波碎石治疗,排出少量结石成分分析示“一水草酸钙”,予口服枸橼酸氢钾钠等治疗。2009年6月复查尿蛋白阴性,红细胞10/μl,白细胞500/μl,Hb 135 g/L,SCr 61 μmol/L,血钙2.33 mmol/L,尿培养阴性,诊断“海绵肾、双肾多发结石”。7月7日行左肾经皮肾镜检查,术中见集合管内被大小不等淡黄色结石充满,最大直径约1 cm,结石与集合管黏膜黏连紧密,结石含基质成分较多,不易破碎,破碎并清除部分较大结石后肾镜进入集合系统,集合系统未见游离结石,肾下盏一个肾乳头开口隐约可见黄褐色结石。2009年7月至2014年2月长期服用枸橼酸氢钾钠治疗,期间喜大量饮水,夜尿次数增多,无腰痛、乏力等不适,未定期随访和复查。2014年2月患者出现明显尿频,排尿1次/h,夜尿5~6次/晚,无尿急、尿痛、发热、腰痛,至我科门诊查尿蛋白定量1.49 g/24h(尿量3 800 ml),尿沉渣红细胞计数(RBC)35万/ml,多形型,白细胞>100/HP,SCr 187 μmol/L,血钾3.44 mmol/L,总二氧化碳18.4 mmol/L,B超 左肾/右肾长径100 mm/95 mm,双肾见数个强回声光团,左肾较大的约16 mm,右肾较大的约16 mm,伴后声影。予枸橼酸氢钾钠颗粒、莫西沙星片及滋肾丸等治疗。4月复查尿蛋白定量2.12 g/24h,SCr 197 μmol/L,尿频症状较前无改善。2014-08-21于某儿童医院查SCr 271.7 μmol/L,C反应蛋白、降钙素原升高,尿白细胞150~200/HP,予抗感染及尿毒清、百令胶囊、开同等治疗,症状无改善,9月30日就诊于我院儿科,复查SCr达805 μmol/L,总二氧化碳12 mmol/L,Hb 94 g/L,予连续性血液净化(CRRT)等治疗。因患者年龄小,外周血管条件差,为行腹膜透析(PD)转入我科,尿量约1 200 ml/d。
既往史 患儿自幼易口渴,饮水量多、尿量多,夜尿次数多。否认肝炎、结核等传染病史,否认“高血压”等病史,否认外伤、输血史。
个人史 无特殊。
家族史 父亲患肾结石,为单个小结石,母亲体健,父母非近亲婚配,1弟弟2岁体健,否认家族中同样疾病患者,否认家族性遗传病史。
体格检查 体温36.7℃,血压116/64 mmHg,身高156 cm,体重47 kg,体质量指数(BMI) 19.31 kg/m2,发育正常,营养中等,贫血貌,全身浅表淋巴结无肿大。心、肺、腹未见异常。季肋点、上输尿管点、中输尿管点、肋脊点和肋腰点无压痛。肋脊角无叩击痛。双下肢无水肿。
实验室检查
尿液 蛋白定量1.16 g/24h,RBC 2万/ml(多形型),尿白细胞>100/HP;α2巨球蛋白(α2-MG)3 mg/L,C3 2 mg/L,RBP 32.7 mg/L,NAG 20.9 U/(g·Cr),Lyso 37.7 mg/L,尿糖阴性。pH 6.5。
血常规 Hb 80 g/L,WBC 6.5×109/L,N/L 57.4%/22%,血小板计数 149×109/L,网织红比例 1.2%。
血生化 白蛋白37.3 g/L,球蛋白30.3 g/L,尿素氮27.8 mmol/L,SCr 904 μmol/L,尿酸431 μmol/L,总二氧化碳22.2 mmol/L,碱性磷酸酶83 U/L,总胆固醇3.4 mmol/L,三酰甘油0.7 mmol/L,电解质正常,钙2.01 mmol/L,磷2.16 mmol/L,估算的肾小球滤过率(eGFR) 8.39 ml/(min·1.73m2)(CKD-EPI公式)。C反应蛋白 19.8 mg/L。空腹血糖4.37 mmol/L。
凝血功能 凝血酶原时间14.1s,凝血酶时间 9s,活化的部分凝血活酶33.6s,纤维蛋白原4.74 g/L。
其他 免疫球蛋白IgG 13.7 g/L,IgA 2.68 g/L,IgM 0.668 g/L,IgE 92.1 IU/ml,类风湿因子(RF)<20 IU/ml。铁8 μmol/L,总铁结合力31 μmol/L,未饱和铁结合力23 μmol/L,转铁蛋白饱和度25.8%。
骨代谢 iPTH 135.5 pg/ml。髂前上棘骨活检:骨髓轻度增生,未见沉积物。
辅助检查
腹部X片 见两肾区见多发结节样高密度影。腹部CT(平扫):两侧肾实质密度正常;双侧肾盏区弥漫分布粟粒样高密度影,呈融合性改变,左肾盏呈囊状扩张积液。肾周脂肪间隙清晰,近段输尿管无明显扩张(图1)。
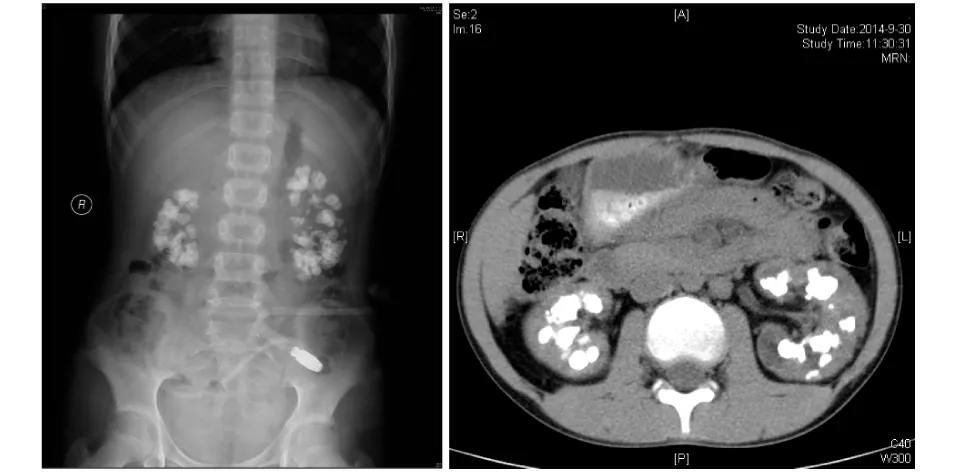
图1 腹部平片及腹部CT见双肾多发结石
双肾B超 LK:左肾/右肾101×36×50/92×41×46 mm,皮质厚度变薄,皮质回声增强,双肾见数个强回声光团,左肾较大的约17 mm,右肾较大的约18 mm,伴后声影。双肾肾锥体回声增强,沿集合系统排列规则。双肾轮廓欠规则,包膜连续完整,结构欠清楚。左肾内局部肾盂肾盏扩张。
心脏超声 室间隔厚度8 mm,左心室内径40 mm,左室后壁厚度8 mm。LVEF:61%。
其他 胸片、心电图正常。
诊断分析 少年男性患者,临床突出表现为双肾多发结石,继而逐渐出现肾功能不全直至终末期肾病(ESRD)。需与以下疾病相鉴别。
髓质海绵肾(MSK) MSK是一种先天性、可能具有遗传倾向的良性肾髓质囊性病变,其特征为肾锥体部的集合管和乳头管先天性呈梭形或小囊状扩张,病变仅限于肾髓质锥体部顶端,位于肾小盏周围,形成多数大小不一的小囊,而肾盂、肾盏、输尿管发育正常。囊肿直径可<1 mm或>6 mm,但>10 mm的囊肿比较少见[1]。MSK可累及单侧或双侧肾脏,影响全部或部分乳头,同时集合管扩张迂曲,尿流不畅,该段尿液中成石成分浓度高,易继发结石和感染。集合管内形成结石时称为海绵肾结石,约占MSK的40%~ 60%。但MSK一般仅累及髓质,预后较好很少发生肾衰竭,更不会累及多个器官,且很少有泌尿系统结石家族史。本例患者未成年即发生肾衰竭,不符合MSK的特点。
甲状旁腺功能亢进、特发性高钙尿症、高钙血症、肾小管酸中毒等疾病继发双肾结石 患者血钙、PTH均正常,可排除上述疾病。
原发性高草酸尿症(PH) 该患者年幼起病,很快进入ESRD,高度考虑该病可能,因无条件行血、尿草酸测定,必须依靠基因检测、骨活检等进一步明确。该患者骨髓活检未见异常,遂采集患者及其父母的血样送基因检测,患者的基因结果回报示两个基因位点变异:(1) GRHPR[c.862至c.863:缺失TG]和[c.435至c.436:缺失GC]:可能导致常染色体隐性遗传的原发性高草酸尿症2型;(2)PKHD1[c.11740G>A,p.3 914R>X]:该患者检出了PKHD1基因的一个杂合变异,使蛋白的第3 914位氨基酸密码子由精氨酸(Arg,R)密码子变为终止密码子。该变异未见文献报道,也未见SNP数据库收录。此变异使蛋白合成提前终止,可能造成蛋白功能丧失,可导致常染色体隐性遗传的多囊肾病。其父母基因位点分别为[c.862至c.863:缺失TG]变异和[c.435至c.436:缺失GC]变异(图2、3)。

图2 先证者及其父基因突变部分

图3 先证者及其母基因突变部分
治疗经过 结合其临床表现、结石成分分析及基因检测结果确诊为PH2型。入院后维持性PD治疗;为防止草酸钙结石进一步加重,加用枸橼酸钾碱化尿液,口服维生素B6减少草酸生成;出院后续随访患者规律PD治疗中,情况良好。
最后诊断 原发性高草酸尿症2型,高草酸尿症相关肾损害CKD 5D期。
讨 论
本例患者为青少年男性,以双肾结石为临床表现,缓慢起病,曾在多家医院诊治,诊断曾考虑过MSK,但肾功能恶化较快,6年即进展至ESRD才在本科通过基因检测最后确诊。对于该患者延迟诊断主要由于临床医师对PH认识不足而导致误诊和漏诊,同时因未能及时或不恰当治疗使患者过早进入ESRD[2]。对于延误诊治的原因,一方面是PH为少见病,医师对其认识不足,即使患者PH和草酸沉积症的临床表现非常典型,仍未考虑到此病;另一方面,临床医师对于“双肾结石”的原因未能仔细探究原因和鉴别。该患者在疾病早期即发现肾结石,同时做过结石成分分析为一水草酸钙结石,如临床医师提高警惕及早做基因检测可确诊。
PH是一种常染色体隐性遗传病。从分子水平可将其分为PH1、PH2和PH3三种类型。PH1是一种遗传性过氧化物酶体疾病,由AGXT基因突变引起,导致维生素B6依赖的肝脏特异的AGT缺失或功能异常[3]。PH2是由GRHPR基因突变引起,主要是D-甘油酸脱氢酶遗传缺陷。正常情况下,羟基丙酮酸盐经该酶催化生成D-甘油酸。该酶缺陷时此反应被乳酸脱氢酶(LDH)取代,生成L-甘油酸。LDH在催化乙醛酸盐生成大量草酸的同时,也使羟基丙酮酸盐代谢产生大量L-甘油酸,出现高草酸尿伴L-甘油酸尿[4]。PH3是由HOGA1基因突变引起,导致线粒体4-羟基-2-酮戊二酸醛缩酶功能异常[5]。三种类型的PH均导致肾脏草酸钙结石病,其中PH1最为常见,约占PH的80%[6]。
三种类型PH的共同临床特征是多在儿童期发病,基因突变导致肝脏乙醛酸盐和草酸盐代谢异常,内源性草酸盐产生过多并从肾脏排泄,导致显著高草酸尿、早期而反复发生的泌尿系统结石(草酸钙结石,X线平片下结石不透光)、持续进展的肾钙质沉着症和进行性肾功能恶化,常有肾结石家族史。草酸还可沉积于肾脏以外的组织器官(如骨骼、关节、心脏、眼睛、皮肤、血管等),病理特征是草酸结晶在组织内(如骨骼、肾小管管腔内)广泛沉积,周围可见反应性肉芽肿围绕。偏光显微镜见折光的晶状物质。
PH1的主要临床特征为多发性和复发性肾脏结石和肾钙质沉着症[7],约有50%患者在儿童期起病,15岁前进展为ESRD。辅助检查有助于PH1的诊断。(1)血、尿草酸测定:在疾病早期无症状时尿草酸排出增加[5],当GFR<60 ml/(min·1.73m2)时,血浆草酸浓度才升高[8];(2)结石成分分析:PH1的结石成分95%为一水草酸钙[6];(3)影像学检查:腹部CT、X线和B超均可发现双肾多发性结石,CT还能发现肾髓质钙质沉着症[6],而X线和B超不能发现肾钙质沉着症;(4)基因分析:诊断PH1的重要和有效的手段[9];(5)肝脏穿刺活检:PH1诊断的金标准[9-10]。台湾和香港地区各有1例儿童期起病的PH1报道,大陆地区有4例儿童期起病的PH1报道,但均未进行AGXT基因检测[11-15]。李国民等[16]报道1例3岁起病10岁进展至ESRD的患者通过AGXT基因分析诊断PH1。国内报告5例肾移植术后复发的患者均在ESRD后行移植肾活检发现小管内广泛草酸沉积而确诊PH1[12,17-20]。
PH2是单基因、常染色体隐性遗传疾病[21],过量的代谢产物通过肾脏排泄导致肾脏损害。这些患者可多年无临床症状,高草酸尿症导致复发性肾结石而较少发生肾钙化,多在出现单侧结石、肾切除术后或在疾病晚期才发现、确诊[22-23]。由于诊断方法受限,截止19世纪90年代仅报道30例的PH2,目前有了更好的认识和分子诊断工具,病例数逐渐增加。虽然PH2患者的临床过程与1型非常相似,但PH2患者较少出现严重的结石及肾钙质沉着,残余肾功能恶化的速度相对缓慢[24]。目前尚无因缺乏GRHPR而导致婴儿期发生肾衰竭的报道。但由于缺乏远期数据,PH2患者仍存在肾功能损害的风险。PH2患者出现严重的肾功能损害时,发生系统性草酸盐血症的风险也随之增加。GRHPR基因位于9号染色体(9p11)和由9个外显子,编码一个328个氨基酸(36 kD)的蛋白。目前为止,已经报道GRHPR基因的15个致病突变,外显子2的一个碱基对缺失G(c.103delg)常见于高加索血统患者(等位基因频率约40%)。突变包括缺失、插入突变、错义突变和无义突变,由此引起的蛋白表达异常或活性丧失。纯合突变与小的缺失和插入发生率高。
常染色体隐性遗传病的阳性诊断依据需要相关基因的纯合或复合杂合有害突变。本例患者的基因分析提示存在GRHPR突变:(1)[c.862至c.863:缺失TG],该变异已被SNP数据库收录(rs180177321)。此变异使开放阅读框移码,可能造成蛋白功能丧失;(2)[c.435至c.436:缺失GC],该变异未见文献报道,也未见SNP数据库收录。此变异使开放阅读框移码,可能造成蛋白功能丧失,而其父亲、母亲各具有上述其中的一个突变,据此认为该家系符合常染色体复合杂合突变遗传规律。
治疗PH的措施主要包括饮水、口服维生素B6、饮食控制以及器官移植。区分1型与2型PH是最重要的。如患者被错误分型可能会导致不恰当的治疗。对于PH1患者可口服维生素B6,如未至ESRD且对维生素B6治疗无显著效果,首先考虑器官移植,可采用肝肾联合移植,应在进展至CKD 4期前进行,避免草酸盐在其他器官沉积[5],这与其他原因引起的CKD5期进行肾脏替代治疗不同。不主张单独肾移植。当不能行肝肾联合移植又有透析指证时需选择高效透析,血液透析(HD)、PD或两者的联合均可,而HD对草酸的清除要优于PD[5,8]。而对于PH2患者应用维生素B6并未获益,且到目前为止尚未证实肝移植可改善高草酸尿症,故而这些患者最好仅进行肾移植[25]。本例患者已进展至ESRD,因血管条件不佳只能行PD治疗。
小结:PH起病隐匿,容易与MSK等疾病混淆。这是国内首例通过基因检测确诊的PH2患者。对有结石家族史或幼年起病、肾功能受损,伴有多个系统受累的患者需考虑到PH的可能。对可疑患者应早期行肾活检或骨髓活检寻找草酸沉积的证据,同时可以行基因检测明确诊断。
1 陈国萍,黄奇虎,房仲平,等.超声对海绵肾的诊断价值.中国超声诊断杂志,2005,6 (1):63-64.
2 Jungers P,Joly D,Barbey F,et al. ESRD caused by nephrolithiasis:prevalence,mechanisms,and prevention.Am J Kidney Dis,2004,44(5):799-7805.
3 Hoppe B,Beck BB,Milliner DS.The primary hyperoxalurias.Kidney Int,2009,75(12):1264-1271.
4 Mansell MA.Primary hyperoxaluria type 2.Nephrol Dial Transplant.1995,10(Suppl 8):58-60.
5 Chand AQ,Kaskel FJ.Pediatrics:Timely diagnosis of primary hyperoxaluria type 1.Nat Rev Nephrol,2009,5(12):670-671.
6 Cochat P,Groothoff J.Primary hyperoxaluria type 1:practical and ethical issues.Pediatr Nephrol,2013,28(12):2273-2281.
7 Edvardsson VO,Goldfarb DS,Lieske JC,et al.Hereditary causes of kidney stones and chronic kidney disease.Pediatr Nephrol,2013,28(10):1923-1942.
8 Cochat P,Hulton SA,Acquaviva C,et al. Primary hyperoxaluria Type 1:indications for screening and guidance for diagnosis and treatment.Nephrol Dial Transplant,2012,27(5):1729-1736.
9 Williams EL,Acquaviva C,Amoroso A,et al. Primary hyperoxaluria type 1:update and additional mutation analysis of the AGXT gene.Hum Mutat,2009,30(6):910-917.
10 Hoppe B.Evidence of true genotype-phenotype correlation in primary hyperoxaluria type 1.Kidney Int,2010,77(5):383-385.
11 程震,唐政,刘志红,等.原发性高草酸尿症误诊二例及文献复习.临床误诊误治,2013,(26)2:18-21.
12 周建华,崔雯,王韵琴.儿童原发性高草酸尿症致双肾广泛结石、钙化和肾功能衰竭一例.中华肾脏病杂志,2004,20(1):46.
13 伍汉文,王艳林.原发性高草酸尿症一例.中华儿科杂志,1994,32(2):88.
14 Wong PN,Tong MW,Mak SK,et al.Late-onset primary hyperoxaluria type 1 in a Chinese individual with absent alanine:glyoxylate aminotransferase activity.Chin Med J (Engl),2004,117(12):1889-1890.
15 Wong PN1,Law EL,Tong GM,et al.Diagnosis of primary hyperoxaluria type 1 by determination of peritoneal dialysate glycolic acid using standard organic-acids analysis method.Perit Dial Int,2003,23(Suppl 2):S210-S213.
16 李国民,沈茜,徐虹,等.儿童原发性1型高草酸尿症1例并文献复习.中国循证儿科杂志,2013,12,(8)453-457.
17 陈晓农,陈楠,潘晓霞,等.原发性高草酸尿症.诊断学理论与实践,2003,2(4):310-312.
18 Shang MH,Jun H,Fan Y,et al.Recurrence of primary hyperoxaluria after kidney transplantation:the report of two cases.Chin Med J (Engl),2009,122(22):2794-2797.
19 朱晓峰,张金元,许龙根,等.原发性高草酸尿症导致的移植肾早期肾功能丧失(附1例报告).东南国防医药,2005,7(4):246-248.
20 黄刚,陈立中,王长希,等.儿童原发性高草酸尿症导致移植肾功能丧失一例.中华器官移植杂志,2007,28(6):378.
21 Rumsby G,Williams E,Coulter-Mackie M.Evaluation of mutation screening as a first line test for the diagnosis of the primary hyperoxalurias.Kidney Int,2004,66(3):959-963.
22 Hoppe B,Langman CB.A United States survey on diagnosis,treatment,and outcome of primary hyperoxaluria.Pediatr Nephrol,2003,18(10):986-991.
23 Kemper MJ,Conrad S,Müller-Wiefel DE.Primary hyperoxaluria type 2.Eur J Pediatr,1997,156(7):509-512.
24 Milliner DS,Wilson DM,Smith LH.Phenotypic expression of primary hyperoxaluria:comparative features of types I and II.Kidney Int,2001,59(1):31-36.
25 Hoppe B,Beck BB,Milliner DS.The primary hyperoxalurias.Kidney Int,2009,75(12):1264-1271.
(本文编辑 律 舟 莫 非)
Kidney stone, hyperoxaluria, and renal dysfunction
ZHOUYan,ZHOUYuchao,LIUZhihong,CHENGZhen
NationalClinicalResearchCenterofKidneyDiseases,JinlingHospital,NanjingUniversitySchoolofMedicine,Nanjing210016,China
A case of a 12-year-old boy was diagnosed nephrolithiasis 6 years ago and laboratory analysis conformed calcium oxalate monohydrate as the composition of the stones. His serum creatinine level was found elevated 8 months ago and his renal function deteriorated in recent one month. His father was also diagnosed nephrolithiasis but the stone was quite small. With the help of monogenic disease screen we confirmed the diagnosis of primary hyperoxaluria. In this case we concluded that analysis of stone composition and GRHPR gene are very useful for diagnosis of hyperoxaluria type Ⅱ.
primary hyperoxaluria kidney stone gene diagnosis
南京军区南京总医院肾脏科 国家肾脏疾病临床医学研究中心 全军肾脏病研究所(南京,210016)
2015-05-07
猜你喜欢
中成药(2021年5期)2021-07-21 08:39:02
基层中医药(2020年6期)2020-09-11 06:35:26
现代临床医学(2019年6期)2019-12-07 06:03:50
家庭医学(下半月)(2019年9期)2019-10-12 08:03:56
材料科学与工程学报(2016年4期)2017-01-15 13:35:34
饮食科学(2016年3期)2016-07-04 15:12:40
饮食科学(2016年3期)2016-07-04 15:12:27
医学研究杂志(2015年3期)2015-06-10 06:41:52
食品工业科技(2014年13期)2014-03-11 18:16:53
中国优生优育(2014年8期)2014-01-20 08:26:45
