
Overview of milling techniques for improving the solubility of poorly water-soluble drugs
2015-05-16 02:13:58ZhiHuiLoh,AsimKumarSamanta,PaulWanSiaHeng
Review
Overview of milling techniques for improving the solubility of poorly water-soluble drugs
ARTICLEINFO
Article history∶
Received 10 August 2014
Received in revised form
28 December 2014
Accepted 29 December 2014
Available online 17 February 2015
∶
Drug solubility
Fluid energy milling
Ball milling
Media milling
High pressure homogenization Cryomilling
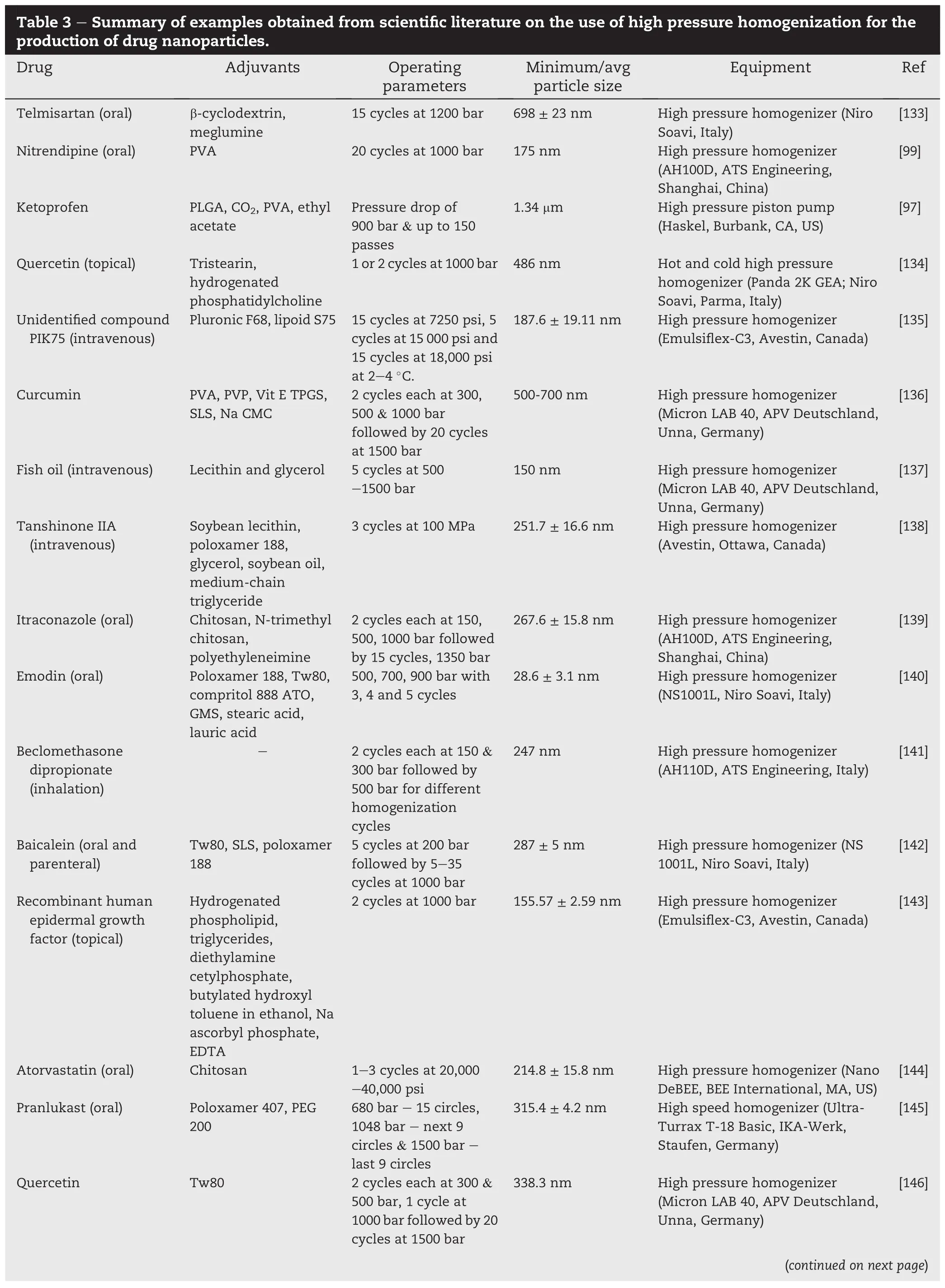
Milling involves the application of mechanical energy to physically break down coarse particles to fi ner ones and is regarded as a“top-down”approach in the production of fi ne particles.Fine drug particulates are especially desired in formulations designed for parenteral,respiratory and transdermal use.Most drugs after crystallization may have to be comminuted and this physical transformation is required to various extents,often to enhance processability or solubility especially for drugs with limited aqueous solubility. The mechanisms by which milling enhances drug dissolution and solubility include alterations in the size,speci fi c surface area and shape of the drug particles as well as millinginduced amorphization and/or structural disordering of the drug crystal(mechanochemical activation).Technology advancements in milling now enable the production of drug micro-and nano-particles on a commercial scale with relative ease.This review will provide a background on milling followed by the introduction of common milling techniques employed for the micronization and nanonization of drugs.Salient information contained in the cited examples are further extracted and summarized for ease of reference by researchers keen on employing these techniques for drug solubility and bioavailability enhancement.
©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/ licenses/by-nc-nd/4.0/).
1. Introduction
The process of drug dissolution is critical to the therapeutic ef fi cacy of a medicinal product regardless of its route of administration.Dissolution involves the transfer of a solid drug into solution in the surrounding physiological fl uid.Therate of dissolution of a drug is affected by factors embodied in the Noyes-Whitney equation[1].The extent to which drug dissolution proceeds under prevailing physiological conditions is governed by its aqueous solubility.Drug solubility is de fi ned as the amount of drug that passes into solution when an equilibrium is established between the drug solute in solution and any excess,un-dissolved drug to produce asaturated solution at a speci fi ed temperature[2].The solubility and dissolution rate of a drug are often positively correlated.The bioavailability of a drug is de fi ned as the rate and extent to which a dissolved drug is absorbed and becomes available at its target site of action[3].The bioavailability of a drug is thus dependent not just on its dissolution and solubility characteristics,but also on its membrane permeability and associated absorption-related degradation.
Currently,the pharmaceutical industry faces considerable challenges associated with the increasing number of poorly water-soluble drugs coming through the drug discovery pipeline[4,5].Despitepromisingpharmacological activity,manyof thesedrugcandidatesfallunderclassIIoftheBiopharmaceutics Classi fi cation System(BCS),characterized by high membrane permeability but low aqueous solubility[6]. These drugs exhibit erratic or incomplete absorption often leading to unsatisfactory drug exposure in vivo and poor bioavailability. Biopharmaceuticals and biotechnologyderived therapeutic agents face similar challenges[7].For BCS class II drugs,the dissolution step is the rate-determining factor in drug absorption.Pharmaceutical scientists are constantly seeking new approaches to facilitate and enhance the solubility and thus dissolution rate of BCS class II drugs. Current strategies employed to improve the apparent solubility of a drug include the use of:(i)co-solvents(e.g.low molecular weight polyethylene glycols and propylene glycol)in combination with water to dissolve the drug;(ii)complexing agents(e.g.cyclodextrins and its derivatives)to form watersoluble inclusion complexes of the drug[8]or(iii)hydrophilic excipients(e.g.polyvinylpyrrolidones and high molecular weight polyethylene glycols)as drug carriers for the preparation of solid dispersions in which the drug is dispersed molecularly or as ultra fi ne crystals[9].Alternatively,the drug molecule may be modi fi ed chemically by the syntheses of suitable pro-drugs[10]or salt forms of the drug that often exhibit greater aqueous solubility than the parent molecule. However,drugprecipitationisacommonthreatfacedbysome of these formulations[11].Precipitation may arise from excess drug coming out of solution when a previously supersaturated drug solution is diluted upon administration.For oral formulations,drugprecipitationmaybetriggeredbythechangingpH environment of the gastro-intestinal tract.To ensure that a drug stays in solution up till the point of absorption,lipids and oilshavebeenemployedasdrugcarriers.Lipidformulationsof drugs basically comprise drugs dispersed,but more often, dissolved,in lipids or oils.These formulations improve drug bioavailability by exploiting the innate lipid digestion and absorption mechanisms in the body.Depending on the chemical nature of the lipid,the formulation may also selfemulsify in the gastro-intestinal tract to facilitate lipid digestion and maximize drug absorption.The advantage of lipid formulations is that the drug is maintained in a solubilized state prior to absorption.A comprehensive reference on oral lipid-based formulations can be found in the book edited by David J.Hauss[12].The key advantages and disadvantages of the different strategies employed to improved drug dissolution and bioavailability are highlighted in Table 1.
2. Milling

Apart from the techniques aforementioned,another strategy employed to improve solubility and ultimately,bioavailability of poorly water-soluble drugs is milling.The terms milling,size reduction,comminution,grinding and pulverization are often used interchangeably.Milling is a unit operation where mechanical energy is applied to physically break down coarse particles to fi ner ones and hence,is regarded as a“top-down”approach in the production of fi ne particles[13].As virtually every drug can be comminuted to fi ne particles regardless of its solubility in aqueous or non-aqueous solvents,the“top--down”approach has wider commercial and industrial applications than the“bottom-up”approach(e.g.precipitation) where fi ne particles are constructed from their dissolved molecular state and suitable solvents/anti-solvents of the drug need to be selected.
Traditionally,milling is carried out to facilitate the extraction of crude drugs or to improve their bulk processing properties.Cutter mills,roller mills,pestle and mortars and runner mills may be employed for this purpose.In these milling operations,the dried crude drug may be cut by sharp blades (cutter mill),impacted by hammers or crushed/compressed by the application of pressure(roller mill,pestle and mortar).As a limited amount of energy is imparted,the milled particles remain relatively coarse.Technological advancements in milling equipment now enable the production of ultra fi ne drug particles down to the micron or even sub-micron dimensions. Griseofulvin,an anti-fungal drug,represents one of the pioneering examples of drugs where solubility and absorption wereenhancedbymilling.Millingofcarbamazepinewasfound to be more effective in enhancing drug dissolution than formulating the drug as a solid dispersion due to polymorphic transformation of the drug(from the β to α form)in the solid dispersion system[14].Other early examples of drugs where milling has resulted in enhanced dissolution include nitrofurantoin,nifedipine,ibuprofen and spironolactone[3].
Milled drug particles are rarely used as it is and are intermediates in the production of pharmaceutical dosage forms.Oftentimes,they are cohesive and exhibit poor fl ow properties,largely due to their higher surface energies compared to their coarser counterparts.To alleviate this problem,inert pharmaceutical excipients or fi llers,e.g.calcium phosphate,lactose,mannitol and other sugars etc.,are often added and mixed with the milled drug particles to improve powder fl ow.Alternatively,the drug particles may be granulated with these fi llers to form granules which typically exhibit improved fl ow properties and content uniformities than thecorrespondingphysicalmixtures.Apartfrom improving fl ow during manufacturing,these fi llers may also serve other functions e.g.modifying drug release,enhancing drug stability and dissolution as well as taste-masking.
3. Mechanisms by which milling improves drug dissolution and solubility
Milled products possess speci fi c physical attributes that contribute to improved drug dissolution and solubility.Milling reduces the size and alters the size distribution of the drug particles.Thesepropertiesmaybemeasuredbylightscattering techniques such as photon correlation spectroscopy(5 μm down to 0.001 μm)and laser diffraction(0.05 μm-2000 μm), respectively[15].Byvirtueoftheirsmallersize,milledparticles possesslargerspeci fi csurface areacomparedtotheirunmilled counterparts.Based on the Noyes-Whitney equation,this is likely to increase the dissolution rate of the milled drug particles if the particles can also be adequately wetted.Milled particles possess higher surface free energies and this,coupled with their thinner diffusion boundary layers[16]further enhance the dissolution rate of the milled drug substance.
Apart from size,milling also alters the surface roughness and shape of particles.This has been demonstrated in many studies on the development of inhalable dry powder formulations.Ithas beenshownthat thesurface properties of milled particles can affect wetting[17]and dissolution behavior[18]. Furthermore,milled particles are rarely isometric or spherical in shape.Compared to particle size,considerably lesser attention has been devoted to the impact of particle shape [19-23]on drug dissolution,solubility and bioavailability although early studies have demonstrated that when particles are platelet-like or possess needle shapes,the shape factors of particles are closely related to their dissolution rates and pro fi les[24-28].Particle shape may be determined by image analysis techniques,laser diffraction[29],scanning electron microscopy,transmission electron microscopy and atomic force microscopy.
In a milling operation,particle size reduction ceases at a practical limit[30]beyond which the material becomes progressively dif fi cult to comminute even when milling time is prolonged.When particle size reduction has reached a critical threshold,the continued transfer of mechanical energy from the mill to the drug substance leads to the accumulation of defects on the drug crystal and disordering of the crystal structure,eventually bringing about the disappearance of the order in the positions of atoms or molecules in the crystal[31]. These defects may manifest throughout the entire crystal resulting in complete amorphization of the drug or be restricted to the crystal surfaces in which case a thin,amorphous(disordered)layer may be formed around a crystalline (ordered)core[32,33].Under these circumstances,the drug is said to be“mechanochemically-transformed”or“activated”by the milling process.Drug amorphization as a result of milling improvestheaqueoussolubilityanddissolutioncharacteristics of the drug.It may also confer additional bene fi ts such as improved compressibility[34].Milling-induced amorphization of drug substances have been reported for piroxicam[35], budesonide[36],naproxen[37]and indomethacin[38]amongst many others.However,the disadvantage of these solid state transformations is that amorphous regions or crystal defects created may be thermodynamically unstable,leading to amorphous-crystalline inter-conversions of the drug during storage,alteration of particle size distribution,speci fi c surface area,chemical and physical reactivity,dissolution or overall performance of the drug product[39].
An approximate measure of the extent of milling-induced drug activation may be obtained experimentally by determining the amorphous content or residual crystallinity before and after milling the drug substance using standard solidstate characterization tools such as x-ray powder diffraction (XRPD),Raman spectroscopy or differential scanning calorimetry.As the information obtained from these different techniques complement each other,a combination of techniques is often desired to fully elucidate the solid-state condition of the milled drug substance.The solubility of manydrugs e.g.griseofulvin[40],chloramphenicol palmitate,indomethacin andphenylbutazonehavebeenenhancedby mechanochemical activation.Comprehensive information on the mechanochemical activation of drugs can be obtained in the review by Boldyrev[31].
4. Milling adjuvants
The micro or nanoparticles produced from milling possess a large surface/interfacial area,increased free energy and decreased thermodynamic stability.These factors promote particle agglomeration.Mechanochemically-activated particle surfaces and amorphous regions generated during milling also increase the surface free energy of the particles,favoring agglomeration.In practice,it has been suggested that particle agglomeration arising from van der Waals'and other forces (e.g.electrostatic forces)become signi fi cant at particles sizes of about 30 μm and below[41].Fine,hydrophobic drug particles less than 5 μm in size are known to be exceptionally prone to agglomeration and this is attributed to the inter-particulate cohesive forces between them.Hence,when milling is prolonged,particle agglomeration may supersede particle fracture and this severely reduces the ef fi ciency of the mill over time.Agglomeration occurring during or after milling reduces the effective surface area of the drug particles,with their resultant dissolution rate and bioavailability,being comparable or even less than their untreated counterparts.It was observed that continued milling of ketoconazole in a cryogenic impact mill led to apparent particle size growth by fi ne particle mechanofusion[42].Aspirin,phenacetin and phenobarbital are known to be prone to the effects of aggregation during particle size reduction.
In most cases,drugs are co-milled together with certain adjuvants to minimize the conditions promoting agglomeration.These adjuvants are inert,non-toxic pharmaceutical excipients that function as a carrier and/or stabilizer of the drug in the milled product.There is considerable variation in the amount of excipient employed,with drug to excipient ratios ranging from 1:3 to 50:1 w/w being reported in the literature[43].Typically,the excipient employed is hydrophilic in nature and notable examples are hydrophilic polymers such as polyvinylpyrrolidone, cellulose ethers, polyethylene glycol,polyvinyl alcohol or poloxamers;surfactants,ionic or non-ionic;inorganic materials like magnesium aluminometasilicate[44]and cyclodextrins.By conferring hydrophilicity to the hydrophobic drug particle surfaces,the added excipient also enhances the wettability,solubility and bioavailability of the poorly water-soluble drug.
The ef fi ciency of a particular stabilizer depends on its potential for interaction with the drug compound.Generally, milling may be conducted with the drug in its dry state(dry milling)or suspended in a liquid medium(wet milling).In dry milling,themechanicalenergyimpartedfostersdrug-excipient interactionsvia van derWaals forcesorhydrogenbonding.The resultant drug-excipient composite particles are often stable, exhibit low tendencies to agglomerate and retain the activated status of the drug[31,34].In wet milling,the addition of surfactants(e.g.sodium lauryl sulfate and polysorbate 80)and polymers(e.g.hydroxypropylmethyl cellulose,hydroxypropyl cellulose,polyvinylpyrrolidone and poloxamer),singly or in combination,helps to minimize the agglomeration of suspendedparticlesviaelectrostaticandstericmechanisms.Steric stabilization is achieved when long chain polymers are adsorbed onto the surfaces of the drug particles,forming a physical barrier that prevents the close approach of the particles.The chain length,molecular weight[45],hydrophobicity[46,47], concentration,shape and surface energy[48]of the polymer will in fl uence the ef fi ciency of adsorption.The method of addingthepolymer(periodicadditionsoradditionatthestartof milling process)also affected its stabilizing properties[49]. Electrostatic stabilization is achieved when charged polymers or ionic surfactants become Adsorbed on the surfaces of the drug particles and lower their apparent charge.Apart from preventingagglomeration,thesestabilizingmoleculesmayalso aid in preventing crystal growth(Ostwald ripening)that could adversely alter the dissolution and bioavailability of the drug suspension after storage.Hydroxypropylmethyl cellulose(3 cps)was found to stabilize and minimize crystal growth in a nanosuspension of an unidenti fi ed drug compound NSV-102 (Novartis Pharma)produced by media milling,an example of wet milling.This was attributed to improved surface coverage owing to its stronger interaction with the drug in comparison with other stabilizers,pluronic F-68,pluronic F-127,sodium lauryl sulfate and polyvinylpyrrolidone K-30,investigated[50]. A screening study of polymers,copolymers,and surfactants has revealed that the stabilizing performance of surfactants to be the best followed by linear synthetic polymers and semisynthetic polymers for the 9 drug compounds(cinnarizine, griseofulvin,indomethacin,mebendazole,naproxen,phenylbutazone,phenytoin,itraconazole and loviride)investigated [51].Commonadjuvantsemployedinthedryandwetmillingof drugs are summarized in Tables 2-4.
Achieving the desired size,shape and activation of drug particles in a milling process often requires extensive optimization of a multitude of process and material-related variables.In terms of processing,a prudent selection of the type of milling equipment is required,followed by the adjustment of the conditions of milling such as the duration of milling, material feed rate and other operational or equipment parameters.Occasionally,a combination of milling techniques may be necessary to achieve the desired outcomes.When such combination techniques are used,numerous processrelated variables need to be adjusted and fi ne-tuned as this allows the unique advantages of each milling technique to be synergistically combined for the desired outcome.Suitable and compatible adjuvants have to be selected to minimize agglomeration,improve wetting,stability and resultant solubility of the milled drug particles.The following sections will provide the background from literature on the common milling techniques employed for size reduction.
5. Milling techniques for the production of microparticles
5.1. Fluid energy milling
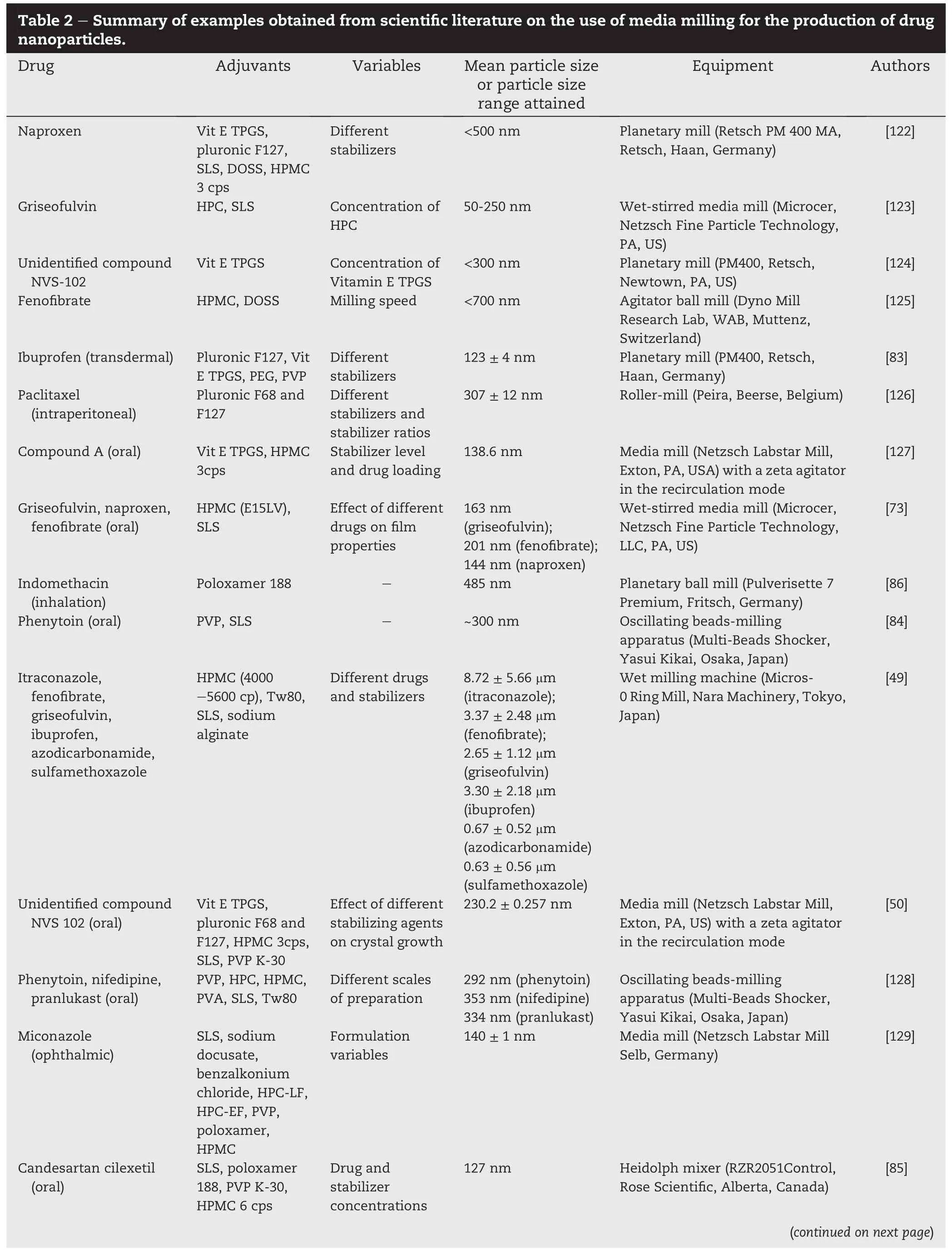
Fluid energy milling,sometimes referred to as air jet milling, effectively reduces the size of drug particles from the range of20-100 μm to less than 10 μm.In this micronization method, high velocity compressed air streams are injected into a chamber where the starting raw materials are fed by a ratecontrolled feeder(Fig.1).As the particles enter the air stream,they are accelerated and caused to collide with each other and the wall of the milling chamber with high velocities. Particle size reduction is brought about by a combination of impact and attrition.Impacts arise from collisions between the rapidly moving particles and particles onto the wall of the milling chamber.Attrition occurs at surfaces of particles as they move rapidly against each other,resulting in shear forces that may break them up.A classi fi er may be integrated into the milling system such that only particles that are suf ficiently fi ne or have acquired dimensions below the prede fi ned cut-off size are entrained in the exhausting air stream and removed from the milling chamber.Classi fi cation may be effected by a spinning wheel classi fi er where the centrifugal force generated by the high spinning speed of the fl uted wheel limits the cut-off size of particles that can accompany the exhausting air through the wheel to the air exhaust outlet.Alternatively,a large tubular shaped milling chamber,circular or oblong,may be used,with colliding air jets at the periphery and the exhaust air leaving centrally, causing a centrifugal classi fi cation system for the milled particles.Longer durations of milling are required when fi ner particles are desired.This process is suitable for meltable materials[52]and drugs that are heat-sensitive.It is also capable ofmanufacturing large quantitiesofpowder continuously.
?
Fluid energy milling has been successfully employed for the micronization of many drugs for the purpose of improving their dissolution and solubility characteristics.Some examples include ibuprofen[53],salbutamol sulfate[54]and fenoterol hydrobromide[55].Drugs are commonly milled on their own although occasionally,co-milling with suitable excipients is carried out.It was reported that fl uid energy milling of a blend of feno fi brate,a poorly water-soluble drug,together with a mixture of hydrophilic excipients resulted in faster drug dissolution rates from a rapidly disintegrating dosage form compared to a powder formulation of identical composition prepared by mixing pre-milled feno fi brate with the excipient mixture[56].This was attributed to the persistence of aggregates of pure,jet-milled feno fi brate which retarded drug dissolution.In an attempt to improve the bioavailability of EMD 57033,a poorly water-soluble calcium sensitizing agent,Vogt et al.[57]found that co-grinding a mixture of EMD 57033 with lactose and hydroxypropylmethyl cellulose using a fl uidenergy millwasmoreeffectivethan micronizingthe drug alone or spray drying a nanosuspension of the drug.Fluid energy milling of ibuprofen together with nanosilica was carried out by Han and co-workers[58].It was found that fl uid energy milling not only decreased the size of drug particles from 102 to<10 μm but also facilitated the coating of nanosilica on the surfaces of the milled drug particles.This surface modi fi cation broughtaboutbymillingreducedparticle agglomeration and improved powder fl ow.
However,the popularity of fl uid energy milling has somewhat dipped following the development of other milling techniques capable of effecting greater extents of size reduction, enabling the production of sub-micron or nanoparticles at commercialscale.Thelowestmean particlediameters achievable by fl uid energy milling is 3-5 μm with size distribution ranging froma few hundred nanometers to about 25μm and a very low fractional content of nanoparticles[59].Nonetheless,this milling technique remains as a benchmark for the evaluation and development of new milling methods and strategies.In the context of drug solubility enhancement, fl uid energy milling may be employed in combination with other particle design techniques(e.g.“bottom-up”approaches such as precipitation,crystallization)to produce drug microparticles with desirable morphological characteristics.Fluid energy milling of ibuprofen was investigated and found generally hard to mill in its dry state due to its ductility and low melting point [53].Inthestudy,ibuprofencrystalsofdifferentsizes(<40μmor 50-250 μm)and morphologies(plate-like and needle-like crystals)were fi rst produced by controlled crystallization.It was reported that the drug could be milled down to less than 5 μm, which is below the reported particle size for brittle-ductile transition of the drug.Furthermore,it was observed that the sizeandmorphologyofthestartingdrugcrystalsin fl uencedthe milling outcome.Compared to plate-like crystals,needle-like crystalsweremoresusceptibletomicronization.Milledparticles produced from smaller-sized starting materials also exhibited smaller volume mean diameters compared to their coarser counterparts for both morphologies of ibuprofen particles.The authors explained that with proper control of the crystal attributes of the starting material, fl uid energy milling might be equally,if not more effective than wet milling in reducing the particle size of drugs.Despite it being a micronization technique, fl uid energy milling also plays a signi fi cant role in the development of nanoparticulate drug delivery systems.Drug microparticles produced from fl uid-energy milling may be subjected to a subsequent milling process to attain particles in nanoscale dimensions.In a study,horseradish peroxidase enzyme as a model drug was loaded in a suitable polymer matrix and pre-micronized using a fl uid energy mill[60].The protein-polymer microparticles were then subjected to a nanonization process(high pressure homogenization)to produce stable,protein-loaded nanoparticles with controlled drug release properties.
?
5.2. Ball milling
Ballmillingisanotherpopularsizereductiontechniqueusedfor the production of microparticles,especially in research laboratories.Fundamentally,a ball mill comprises a vessel or vial fi lled withballs,or rods,constructedfroma variety ofmaterials such as ceramic,agate,silicon nitride,sintered corundum,zirconia,chrome steel,Cr-Ni steel,tungsten carbide or plastic polyamide(Fig.2).The material to bemilled isplaced inside the vessel,whichismadetorotateorvibrateataparticularspeedor frequency.The movement of the vessel causes the balls to cascade or move in a particular pattern,colliding with each other and with the opposing inner wall of the vessel.Size reduction of the drug particles is effected from the impact they receivefromtheballs aswellas attritiveforcesarising from the movement of the balls relative to each other[61,62].
The quantities of the balls and starting material determine the extent of fi ll of the vessel and the intensity of the milling process.Typically,the vessel is fi lled by the balls and starting material to 50%and 25%of the total volume of the vessel, respectively,although variations exist in the literature.In the case of a rotating vessel,rotation is usually carried out at 50-85%ofthe criticalspeed,de fi nedas thespeedatwhichthe balls cease to cascade owing to the centrifugal force imparted by the rotating vessel.The critical speed may be estimated based on the following relationship[63]:
The use of ball milling as a micronization technique for enhancing drug solubility is well supported by literature dating as far back as the 1970s.Apart from its comminution function,ball milling also serves as an intensive mixing technique capable of producing co-ground drug-excipient mixturescomprising amorphous drugformsintimately mixed with suitable hydrophilic excipients at the molecular level. This interaction between the drug and hydrophilic excipient enhances the wetting and dissolution of the drug.Studies on the vibrational ball milling of griseofulvin and phenytoin have demonstrated that milling these drugs in combination with microcrystalline cellulose resulted in amorphization of the drug,enhancing its dissolution and bioavailability[64,65].Ball milling of ibuprofen and aluminum hydroxide was carried out tofacilitate complex formation between ibuprofen and aluminum hydroxide as well as drug amorphization which enhanced the dissolution of ibuprofen[66].The incorporation of excipients may,in some cases,mitigate milling-induced drug amorphization.It was demonstrated that the co-milling of salbutamol sulfate with crystalline excipients(α-lactose monohydrate,adipic acid,magnesium stearate)in a ball mill was effective in reducing milling-induced amorphization or structural disorder of salbutamol sulfate[67].Ball milling of puredrugmixtures havealsobeeninvestigated.More recently,ball milling of a combination of two BCS Class II drugs,simvastatin and glipizide,resulted in the formation of stable co-amorphous mixtures[68].
Despite the ef fi ciency of ball milling for size reduction or amorphization,it is less amendable to scale up.By a fl owthrough method,with vibrating balls or discs in the milling chamber,milling ef fi ciency can be improved.External jacketing for heat removal allows the mill to be used continuously and will limit the rise in temperature within the milling chamber.
6. Milling techniques for the production of nanoparticles
6.1. Wet milling
Both fl uid energy and ball milling techniques involve size reduction of drug particles in their dry state.The extent of size reduction achievablein these dry millingtechniques is limited to a few micrometers[69].It was reported that when the solubility of a drug is very low,down-sizing it to the micrometer range is insuf fi cient to increase its dissolution rate and gastrointestinal absorption[70].In the last decade,signi fi cant advancements and evolutions in milling processes have enabled the production of submicron-sized(<1 μm)or nanoparticles [43].This process may be termed as nanonization.Nanoparticles,sometimes termed as nanocrystals,are typically 200-500 nm in size[71],and are particularly suited for the formulation of parenteral preparations. Nanoparticles possess signi fi cant advantages over microparticles in enhancing drug solubility.Most notably,the process of nanonization increases not only the surface area and dissolution rate of the drug particles,but also the saturation solubility of the drug.Ordinarily,the saturation solubility of a drug is dependent on the temperature and solvent used for dissolution.When the size of drug particles falls below 1 μm, dissolution pressure increases due to the strong curvature of the particle surface.This leads to an increase in saturation solubility in accordance to the Ostwald-Freundlich and Kelvin equations.Hence,at the nanoparticulate level,the saturation solubility of a drug becomes a function of particle size.The increase in saturation solubility of the drug will increase the concentration gradient for drug diffusion and promote drug absorption.Additionally,it was reported that orally-delivered nanoparticles displayed strong adhesive properties to the mucosal surfaces of the gastro-intestinal tract,arising from the increased van der Waals forces of attraction between the nanoparticles and the gut wall.This would further contribute to the increased absorption and bioavailability of drugs administered as nanoparticles.
Drug nanoparticles are most commonly produced by wet milling.As the name suggests,wet milling involves size reduction of drug particles suspended in a liquid medium that may be aqueous or non-aqueous in nature.Wet milling is particularly suited for potent drugs and drugs which possess high residual moisture contents(>50%moisture)because dry milling may be problematic for drugs of this nature.In wet milling,a drug nanosuspension is produced as the end product although for improved product stability(minimization of Ostwald ripening and possible hydrolytic degradation of drug),patient convenience and the drive towards green or sustainable manufacturing processes,the nanosuspension may subsequently be transformed into a solid dosage form (e.g.tablets and capsules)by granulation,freeze drying and spray drying[43]using suitable excipients or“matrix formers”like mannitol or lactose.The dissolution rate of ezetimibe,a lipid-lowering compound,was improved even after nanocrystals of the drug were tableted without the inclusion of any solubilizing agents like sodium lauryl sulfate[72].Drug nanoparticles may also be incorporated into polymer fi lms [73]orlayeredonsugarbeads[74].Ithasbeendemonstratedin many studies that these“solidi fi cation”processes retain the mechanically-activated status and other desirable physical attributes of the original,milled drug particles.
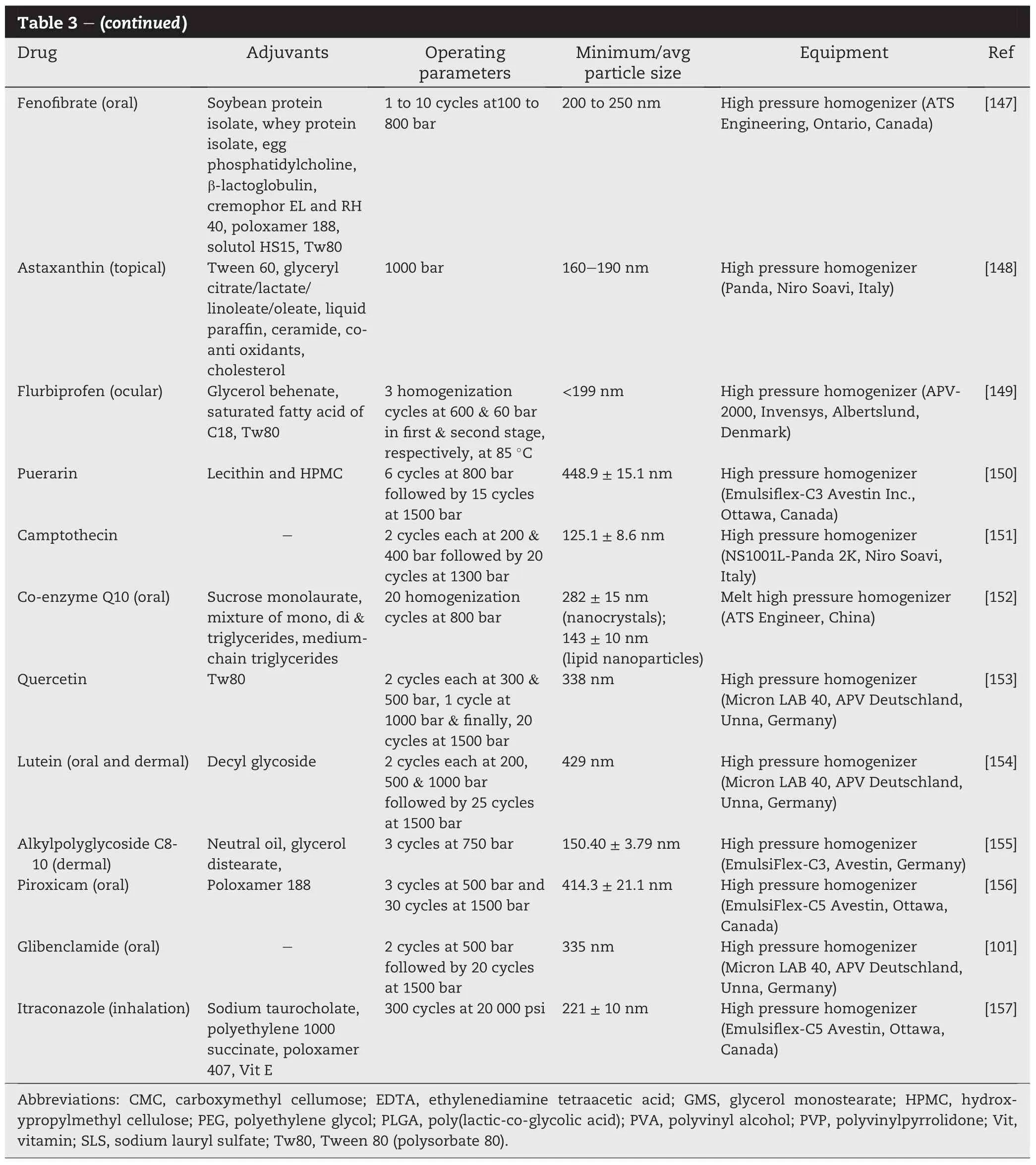
Two common“top-down”approaches for the production of drug nanoparticles are media milling and high pressure (piston-gap)homogenization.These two milling techniques have been the focal point of research in the past decade due to their ease of scale-up,robust processing,economic advantages and acceptance by regulatory authorities.Since 2000, the USFood andDrugAdministrationhas approvednumerous products e.g.Rapamune®(sirolimus,Wyeth),TriCor®(feno fibrate,Abbott)and Megace®ES(megestrol acetate,Par Pharmaceuticals)that have been produced by these techniques [75].Tables 1 and 2 summarize the current research on the application of media milling and high pressure homogenization in particle size reduction and solubility enhancement of speci fi c drugs.
6.2. Media milling
Media milling can be considered a modernized version of the ball mill(Fig.3).This technology, fi rst developed by Liversidge and co-workers[76,77],is a classical wet milling technique wherein a suf fi ciently concentrated dispersion of drug particles in an aqueous or non-aqueous liquid mediumis subjected to a traditional ball milling operation[77,78].The liquid medium prevents adhesion and subsequent compaction of the milled drug particles on the wall of the vessel and/or the surfaces of the milling balls,which is a common occurrence when the drug is milled in its dry state.This improves the yield of nanoparticles.The liquid may also serve additional purposes such as lubrication and coating of the newly-formed particle surfaces through various physicochemicalinteractions like electrostatic and hydrophobic interactions [79,80].
In media milling,mechanical attrition and impaction of the suspended drug particles are brought about by grinding balls,often termed as the milling media,constructed out of a variety of material such as glass(yttrium-stabilized),zirconium oxide,ceramics or highly cross-linked polystyrene resins[78].Pearl balls and beads are commonly used as well in which case the techniques are termed pearl and bead milling, respectively.Unlike ball milling where the whole vessel rotates or oscillate/vibrates whilst in operation,the vessel remains stationary in media milling.Movement of the balls is initiated by a stirring or agitating device,often represented by several discs mounted on a central shaft rotating at high velocities,20 000 rpm and above,within the vessel.For this reason,media milling is sometimes known as“stirred-ball milling”.Media milling is a continuous process wherein the drug suspension is pumped through the milling chamber to effect size reduction of the suspended material.Prior to their exit from the milling chamber,the milled particles pass through a screen that serves to separate the suspended,milled particles from the milling media.Media milling has been employed for particle size reduction of loviride[81],ezetimibe [72],alpha-lipoic acid[82],ibuprofen[83],cinnarizine,naproxen[74],ketoconazole,phenytoin[84]and candesartan cilexetil[85].The majority of these cited studies involve theconversion of the resultant drug nanosuspension into a suitable solid dosage form like dry powders and tablets.Less conventionally,stable nanoparticles of naproxen,feno fi brate and griseofulvin produced from wet-stirred media milling have also been incorporated into hydroxypropylmethyl cellulose polymer fi lms[73].The dissolution rates of the drugs were improved by nanonization and based on x-ray diffraction and Raman spectroscopic analysis,the process of fi lm formationdid not affectdrug crystallinity.Another interesting example is the incorporation of crystalline nanoparticles of indomethacin,preparedbymediamilling,intocoated mannitolmicroparticlesusingan aerosol fl ow reactor method.The nanostructured microparticles produced exhibited rapid dissolution properties[86].
In recent years,there is a trend towards the use of milling media of much smaller dimensions(<100 μm)to bring about the nanonization of drugs.As it is dif fi cult to separate milling media of this size from the milled products using the conventional screen separator without choking or plugging the screen,centrifugal technology has been employed to effect such separations.The Ultra Apex Mill(Kotobuki Industries)is an example where centrifugal technology has been integrated into the design of the mill to effectively separate the milling media,which can range from 15 to 100 μm,from the milled product.This mill has been successfully employed for the production of nanoparticles of albendazole,danazol and omeprazole as well as probucol[87]for the enhancement of their dissolution and absorption properties.
A major drawback of media milling is the erosion of the balls arising from the intensive mixing forces in the vessel. Residues of the milling media produced from erosion may result in product contamination[71,88,89],leading to chemical destabilization of the newly-formed particle surfaces and possibly affecting critical product attributes such as particle size and size distribution.Monitoring impurity levels in the fi nal milled product is thus warranted.Contamination problems may be mitigated by a prudent choice of the materials such as erosion-resistant polymers and ceramics,used in the construct of the milling media as well as other key equipment components(e.g.inner walls of milling chamber and stirring device)in contact with the milled product.Optimization of key process parameters like stirring speed and in particular, milling time also contributes to reducing the likelihood of erosion.This is because milling durationsof up to several days are not uncommon in media milling[77]and such long milling durations are likely to promote erosion of the milling media. Commercially,media milling is exempli fi ed by the Nano-Crystal®technology from Elan Pharmaceutical Technologies. To date,NanoCrystal®-based products have been approved by worldwide regulatory agencies including the US,Canada,EU and Japan.
6.3. High pressure homogenization
The production of nanosuspensions using high pressure or piston-gap homogenization was fi rst developed by Muller et al.in 1994[90].It is a high energy process in which size reduction of drug particles is achieved by repeatedly cycling, to 200 plus cycles,with the aid of a piston,a drug suspension through a very thin gap at high velocity,around 500 m/s,and pressure,1000-1500 bars(Fig.4).The width of the gap,which generally falls within the range of 5-20 μm,may be adjusted according to the viscosity of the suspension and the applied pressure.Pre-micronization of the starting materials using a process like fl uid-energy milling may be necessary prior to homogenization.This is to minimize clogging of the homogenization gap and to reduce milling time.When the suspension is forced through the gap at a high fl ow rate,the static pressure exerted on the liquid falls below the vapor pressure of the liquid at the prevailing temperature(Bernoulli's equation).As a result,the liquid boils and gas bubbles are formed which collapse when the liquid exits from the gap and normal pressure is resumed.The powerful cavitation forces arising from the formation and collapse of the gas bubbles,coupled with a shearing effect,bring about nanonization of the drug microparticles[91-93].The extent of subdivision of the nanoparticles depends on the pressure applied as well as the number of passes or homogenization cycles the drug suspension is subjected to during the process.The mechanical properties(hardness)of the drug particles also affect the milling outcome,with mechanically softer drug particles achieving a lower(200-300 nm)nanometer size range eventually[7].High pressure homogenization is a high energy process.In the course of milling,the drug particles are exposed to a power density of up to 1013 W/m3which is comparable to power densities observed in nuclear power stations.The highenergyimpartedmechanically activatesthe drug particles potentially leading to partial or complete amorphization of the drug[94].High pressure homogenizers are available in different capacities,ranging from few tens of milliliters for laboratory use to a few thousand liters for large scale production.The process is robust and may easily be adapted for aseptic production of parenteral drug nanosuspensions.High pressure homogenization is employed for the production of parenteral nutrition emulsions(e.g.Lipofundin®and Intralipid®).High pressure homogenization also enables continuous processing and unlike media milling, contamination problem from milling equipment is much less. In the last decade or so,extensive research has been carried out on the application of high pressure homogenization for particle size reduction and solubility enhancement of drugs. Table 2 summarizes the research reports in the area over the recent years.Commercially,high pressure homogenization is exempli fi ed by the Dissocubes™technology which has been owned by SkyePharma PLC since 1999.Nanosuspensions prepared using Dissocubes™are water-based and are characterized by well-de fi ned particle sizes and narrow particle size distributions,with few particles exceeding 5 μm in size [95].Many different drugs have been processed by the Dissocubes™ technology resulting in bioavailability enhancement.
In 1999,a variant of the high pressure homogenization process,known as the Nanopure®(pure nanocrystals)technology,was introduced.This technology is owned and developed by Pharmasol GmbH,Berlin.Homogenization in this case is conducted in a non-aqueous dispersion medium, polyethylene glycols,propylene glycol,water-alcoholic mixtures or suitable oils[96],rendering the process suitable for comminuting drugs susceptible to hydrolysis.This technique is also suitable for drugs that are thermolabile.This stems from the option of conducting homogenization under milder conditions such as reduced temperature[97]and in the event where the nanosuspension has to be converted into a solid form,less heat will be required for solvent removal(compared to homogenization using water)thereby preserving drug stability.Supercritical CO2has also been employed as a dispersion medium.In two recentstudies,high pressure homogenization ofphenytoin [98]and ketoprofen [97] dispersed in supercritical CO2was carried out.The use of supercritical CO2enhanced the ef fi ciency of homogenization as the liquid medium could be easily evaporated under ambient conditions,allowing the milled product to be readily recovered in the form of a dry powder.
High pressure homogenization has also been combined with precipitation and this is marketed as the Nanoedge™technology.In this technique,the drug is fi rst dissolved in a water-miscible alcoholic solvent(e.g.methanol,ethanol or isopropanol),then added to water to cause it to precipitate. The precipitated particles are subsequently homogenized. The homogenization step is purported to reduce the size and size distribution of the precipitated particles,thereby minimizing the likelihood of crystal growth and improving the stability of the nanosuspension during storage.Precipitation, in tandem with high pressure homogenization,has been successfully applied for the preparation of nitrendipine nanocrystals to improve drug bioavailability.Chitosan,which was used subsequently to modify the surfaces of the nanocrystals,furtherenhanced drug bioavailability without bringing about any measurable increase in the size of the original nanocrystals[99].
6.4. Cryogenic milling
In principle,size reduction begins when the externallyapplied stress induces suf fi cient strain within the particles and causes the formation of cracks.The cracks are then propagated through lines of weaknesses in the material,with new cracks being initiated and perpetuated along the way at other discontinuities.A cascade effect occurs and material fracture results.The mechanical property such as hardness or elasticity of a drug is likely to affect the ease of crack initiation and propagation during milling.Logically,harder materials require greater energy input to effect particle size reduction as they do not yield to the externally applied stress as readily as softer materials.However,the brittleness or plasticity of the material also plays a critical role in rendering the ease of particle size reduction.It is easier to comminute hard and brittle materials,such as chalk,than soft and viscoelastic materials,such as rubber,waxes or natural gums.Instead ofbrittle fracture,more plasticmaterialsare capable of absorbing large amounts of energy through viscoelastic deformation without crack initiation or propagation.At ambient conditions,these materials resist fracture or may even melt(for waxy materials)when exposed to the heat generated during milling.Drug substances that exhibit such characteristics are not likely to be amenable to the conventional dry or wet milling methods aforementioned.However, there had been few studies on the milling of soft and highly plastic pharmaceutical materials[52].
Cryogenic milling or cryomilling in short,is a size reduction method specially catered to soft,elastic/plastic,nonbrittle and thermolabile materials.Cryomilling may be carried out either by fi rst freezing the materials in liquid nitrogen (-100 to-150°C)prior to milling or by milling the materials under cryogenic conditions,i.e.,in the presence of liquid nitrogen(Fig.5).Exposure to liquid nitrogen results in the“embrittlement”of the material which facilitates crack propagation,reducing the speci fi c energy required for milling[100] and potentially shortening the milling duration.Although freeze drying performs a similar function and renders a material brittle and porous,by virtue of the fact that it is a drying process implies that it is more suitable for liquids or solid materials containing higher residual moisture contents.Salazar et al.[101]studied media milling and high pressure homogenization of glibenclamide that was fi rst pre-treated by freeze-drying.Freeze-drying rendered the drug brittle and porous,facilitating the subsequent milling process.This combination approach reduced milling time and improved milling ef fi ciency.
Cryomilling enables the production of both micron and nano-sized particles but it has not been widely adopted in the pharmaceutical industry for the milling of drugs.Sugimoto et al.[102]studied the cryogenic co-milling of phenytoin and polyvinylpyrrolidone as a means to improve the dissolution rate of the drug.Phenytoin was dispersed in liquid nitrogen and subjected to media milling using beads constructed out of zirconium or dry ice.Spontaneous sublimation of the dry ice beads together with liquid nitrogen at ambient condition enabled the recovery of a high yield(85-90%)of dry phenytoin nanoparticles,since material loss arising from adhesion to the surfaces of the milling media was of no consequence.In another study,pluronic F-68,a soft and meltable material that may be used both as an excipient and active ingredient in inhalable dry powder formulations[103], was subjected to a micro-ball milling technique using stainless steel ball bearings as the grinding agent.Ball milling was carried out in the presence of liquid nitrogen vapor.Apart from chilling the chamber,the liquid nitrogen also prevented plastic deformation of the material and improved the particle fracture process[52].
Cryomilling is advantageous in that it minimizes the degradation of thermolabile drug substances and loss of volatile drug compounds.It also reduces the risk of explosion, oxidation of formulation constituents and particle aggregation during the milling[104].The latter stems from the low surface tensionandviscosityofliquidnitrogenwhichallowsit to penetrate into inter and intra-particulate void spaces and micropores of the particles,forming a physical barrier which prevents particle agglomeration[105].Cryomilling decreases the effect of temperature-induced changes during milling. Jayasankar et al[106],in a study on co-crystal formation betweencarbamazepineandsaccharin,reportedthatcogrinding the drug and excipient under cryogenic conditions was necessary to prevent the reaction from proceeding through the melt phase which commonly occurs when cogrinding is carried out at ambient conditions.Cryogenic cogrinding also led to higher levels of amorphization than cogrinding at room temperature.These results are echoed in another study involving the ball milling of 3 different crystalline forms of piroxicam[107].Differing extents of drug amorphization was achieved by ball milling the drug under different temperature conditions(ambient and cryogenic conditions),with cryogenic ball milling beingmore effective in inducing drug amorphization.This is because milling at cryogenic temperatures effectively‘traps’the milled material in its amorphous state by removing the thermal energy required for re-crystallization to occur[108].In this regard, cryogenic milling is advantageous as it enables the production of amorphous material without the deleterious effects of solvents or heating[61].Crowley and Zogra fi[109]studied the cryogenic grinding of 5 crystal forms of indomethacin and found that amorphization occurred for one of the solvates (indomethacin methanolate)and all the three polymorphs(γ, α,and δ)studied.Recently,γ-indomethacin was subjected to cryomillingand itwasreported thattheamorphousindomethacin produced exhibited enhanced dissolution rates which was positively related to the duration of cryomilling [38].The same group of researchers had also evaluated the physical stability of the cryomilled amorphous indomethacin samples by measuring the time required for the drug to recrystallize on storage[110].Cryogenic grinding was successfully used to convert crystalline glibenclamide to its amorphous form,averting possible chemical degradation during theprocess.Thecrystalline-amorphousconversionwas shown to be connected with the amide-imidic acid tautomerism of glibenclamide[111].Chieng and co-workers[112] investigated the effect of cryomilling on 2 polymorphic forms of ranitidine hydrochloride and evaluated the physical stability of the milled amorphous drug under different storage conditions.
High impact cryomilling thus enables the production of completely amorphous drugs that may otherwise be dif fi cult to obtain by milling at room temperature.However,drug amorphization may not always be advantageous from a stability point of view.In a recent study on the cryomilling of furosemide,it was reported that the duration of cryomilling andresultant drugamorphization werefactorsresponsiblefor the chemical decomposition of the drug[113].Drug amorphization may not occur in all cases.In a study by Niwa et al. [114],the nanocrystals of phenytoin,ibuprofen and salbutamol sulfate produced from an optimized cryomilling process retained their crystalline character.This was explained by the mild processing conditions that prevailed during cryomilling. Feng et al.[115]reported that cryogenic milling of griseofulvin led to a reduction in drug crystallinity due to the increase of crystal defects,rather than the formation of amorphous drug. A body of research on the use of cryomilling to process molecular materials,including model pharmaceutical compounds,has been carried out by Willart and Descamps[108]. Table 3 summarizes the latest research fi ndings on the use of cryomilling for particle size reduction and solubility enhancement of drugs.
7. Process analytical technology(PAT)in milling
In 2004,the Food and Drug Administration(FDA)initiated the quality by design(QbD)concept,in which process analytics are embedded to monitor,in real time,critical process and product attributes rapidly and non-destructively.The aim of the PAT initiative is to diminish the reliance on end product testing to ascertain product quality,improve process understanding as well as develop intelligent sensing and responsive manufacturing processes.It represents the agency's move to a science-based approach to pharmaceutical manufacturing.In line with this initiative,there is a need to implement process analytical tools to achieve better understanding of the particle phenomena during milling.As aforementioned,the size and shape of milled particles can affect the dissolution properties, resultant bioavailability and storage stability of the formulated product.Hence,it is critical to monitor and track the evolution of particle size and shape during milling.Laser diffraction has become the method of choice for particle size measurement during milling due to its versatility,rapid and reproducible particle sizing[116].It uses light-scattering measurements to calculate the volume-based particle size distribution.In the last decade,alternative techniques to characterize particle size distribution based on focused beam re fl ectancemeasurement(FBRM),ultrasonicattenuation spectroscopy(UAS),phase Doppler method(PDA),spatial fi ltering technique(SFT)and shadow Doppler velocimetry (SDV)have surfaced.FBRM uses a focused beam of laser light that scans across a particle passing in front of the probe window to measure a chord length distribution.UAS measuresthevolume-based particlesizedistribution from extinction spectra using the fundamental equations of mass, momentum and energy balance describing the interaction between an ultrasonic wave and suspended particle.The PDA is based upon the principles of light scattering interferometry and measurements are made using the same optical probe as for laser Doppler velocimetry(LDV).SFT uses the measuring principles of the fi ber optical spatial fi ltering velocimetry(SFV) and the fi ber optical spot scanning(FSS)in order to determine simultaneously the size and the velocity of particles.SFV is a method ofthevelocitydeterminationofanobjectby observing the object through a spatial fi lter in front of a receiver.FSS is an addition to the SFV to observe the shadow image of a moving particle through a single optical fi ber with a small diameter.SDV is based on the imaging of a conventional LDV probe volume onto a linear photodiode array.
Particle size measurements can be made in-line or on-line. In in-line particle sizing,a probe is directly inserted into the process stream for measurements.However,the main process streamoftenoperatesathighparticle fl owrates.Itisthereforea common method to measure in a side-stream that can be isolated from the main process fl ow.Another option is the application of a dilution step between the particle stream and the measuringdevice.Inon-lineparticlesizing,asampleisdiverted from the manufacturing process stream for measurement,to circumvent the high particle fl ow rate in the main stream.
As particle shape along with size are directly related to the product quality and performance,the use of a single parameter to describe the physical dimension of particles is often insuf fi cient,especially for non-spherical(rod or plate-like) particles.Therefore,in-line or on-line particle sizing together with shape measurement can play a crucial role in controlling milling processes.However,little attention has been given to real-time particle shape measurement due to the lack of available technologies.Laser Doppler methods have been employed to analyze particle shape,but only with limited success[29,117,118].Dynamic image analysis(DIA)is still the most commonly used technique for particle shape characterization.However,manual operation was required for sample preparation,measurement and data analysis due to low level of automation.Recent advancements in technologies of high speed cameras and computers have increased the level of automation which enabled the measurement of two dimensional images of dynamic particles[119].DIA was speci fi cally suitable in sizing non-spherical particles[119,120]. However,the use of the DIA systems have been mostly limited to wet analysis,where particles are suspended in a liquid medium to allow easy control of particle fl ow rate and thus reduction of motion blur during image acquisition. Recently,SympatecInc.(Clausthal-Zellerfeld,Germany)commercialized a DIA system(QICPIC)that is capable of capturing images of dry powder particles in a rapidly-moving air stream.Another introduction of DIA system is the noninvasive,real-time 3D particle characterizer capable of giving live particle size and shape information for coarser particles is the Eyecon™(Innopharma Labs,Dublin,Ireland)which uses a unique illumination technique to assist in detection of particle boundaries.The different PAT approaches in milling are summarized in Table 5.

8. Conclusion
A feature of advancement in the pharmaceutical industry is the increased use of materials,particularly drugs,in their fi nest state of subdivision to enhance dissolution,solubility and bioavailability.A myriad of milling techniques and equipment are now available for particle size reduction of drugs,with many capable of scaling up and adaptable to consistent and continuous manufacturing.The application of PAT tools in milling also improves process understanding as well as facilitates the monitoring and control of the particle size reduction process.This enables the production of fi ne particulates with predictable and controllable physicochemical characteristics.There are other aspects of milling that are not within the scope of this review but nonetheless warrant considerable attention.These include the possible cellular toxicities of drug nanoparticles as well as the increased risks of chemical degradation,polymorphism and loss of the activities of drugs as a result of them being milled to a fi nelydivided state.Despite recent progress and innovations in the artof milling,mucheffortis stillneededto improvetheenergy ef fi ciencies of milling operations particularly those designed for ultra fi ne grinding.Even in the most ef fi cient mills,as little as 2%of the total energy consumption may be channeled to effect particle size reduction,with the remainder being lost via elastic/plastic deformation of particles,inter-particulate and particle-machine friction,heat,sound and vibration [121].The use of milling as a means to engineer and produce fi ne,surface-modi fi ed particles also represents an exciting area of research wherein the function of milling extends beyond size reduction and offers a more holistic and integrated approach to particle design.
REFERENCES
[1]Noyes AA,Whitney WR.The rate of solution of solid substances in their own solutions.J Am Chem Soc 1897;19:930-934.
[2]Aulton M.Dissolution and solubility.In:Aulton ME,editor. Pharmaceutics:the science of dosage form design.2nd ed. London:Churchill Livingstone;2002.p.15-32.
[3]Ashford M.Assessment of biopharmaceutical properties.In: Aulton M,editor.Pharmaceutics:the science of dosage form design.2nd ed.London:Churchill Livingstone;2002. p.253-273.
[4]Lipinski CA,Lombardo F,Dominy BW,et al.Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 1997;23:3-25.
[5]Lipinski CA.Drug-like properties and the causes of poor solubility and poor permeability.J Pharmacol Toxicol 2000;44:235-249.
[6]Amidon GL,Lennernas H,Shah VP,et al.A theoretical basis for a biopharmaceutic drug classi fi cation:the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 1995;12:413-420.
[7]Muller RH,Keck CM.Challenges and solutions for the delivery of biotech drugs-a review of drug nanocrystal technology and lipid nanoparticles.J Biotechnol 2004;113:151-170.
[8]Loftsson T,Brewster ME.Pharmaceutical applications of cyclodextrins.1.Drug solubilization and stabilization. J Pharm Sci 1996;85:1017-1025.
[9]Serajuddin ATM.Solid dispersion of poorly water-soluble drugs:early promises,subsequent problems,and recent breakthroughs.J Pharm Sci 1999;88:1058-1066.
[10]Ettmayer P,Amidon GL,Clement B,et al.Lessons learned from marketed and investigational prodrugs.J Med Chem 2004;47:2393-2404.
[11]Johnson JLH,He Y,Yalkowsky SH.Prediction of precipitation-induced phlebitis:a statistical validation of an in vitro model.J Pharm Sci 2003;92:1574-1581.
[12]Hauss DJ.Oral lipid-based formulations:enhancing the bioavailability of poorly water-soluble drugs.New York: Informa Healthcare;2007.
[13]Rabinow BE.Nanosuspensions in drug delivery.Nat Rev Drug Discov 2004;3:785-796.
[14]Machiste EO,Giunchedi P,Setti M,et al.Characterization of carbamazepine in systems containing a dissolution rate enhancer.Int J Pharm 1995;126:65-72.
[15]Randall CS.Particle size distribution.In:Brittain HG,editor. Physical characterization of pharmaceutical solids.New York:Marcel Dekker Inc;1995.p.157-183.
[16]Bisrat M,Nystro¨m C.Physicochemical aspects of drug release.Viii.The relation between particle size and surface speci fi c dissolution rate in agitated suspensions.Int J Pharm 1988;47:223-231.
[17]Ho R,Dilworth SE,Williams DR,et al.Role of surface chemistry and energetics in high shear wet granulation.Ind Eng Chem Res 2011;50:9642-9649.
[18]Danesh A,Connell SD,Davies MC,et al.An in situ dissolution study of aspirin crystal planes(100)and(001)by atomic force microscopy.Pharm Res 2001;18:299-303.
[19]Massol-Chaudeur S,Berthiaux H,Dodds JA.Experimental study of the mixing kinetics of binary pharmaceutical powder.Chem Eng Sci 2002;57:4053-4065.
[20]Mullarney MP,Hancock BC,Carlson GT,et al.The powder fl ow and compact mechanical properties of sucrose and three high-intensity sweeteners used in chewable tablets. Int J Pharm 2003;257:227-236.
[21]Shah KR,Badawy SIF,Szemraj MM,et al.Assessment of segregation potential of powder blends.Pharm Dev Technol 2007;12:457-462.
[22]Swaminathan V,Kildsig DO.Polydisperse powder mixtures: effect of particle size and shape on mixture stability.Drug Dev Ind Pharm 2002;28:41-48.
[23]Yin SX,Franchini M,Chen J,et al.Bioavailability enhancement of a cox-2 inhibitor,bms-347070,from a nanocrystalline dispersion prepared by spray-drying. J Pharm Sci 2005;94:1598-1607.
[24]Dali MV,Carstensen JT.Effect of change in shape factor of a single crystal on its dissolution behavior.Pharm Res 1996;13:155-162.
[25]Fini A,Fazio G,Fernaˊndez-Hervaˊs MJ,et al.In fl uence of crystallization solvent and dissolution behaviour for a diclofenac salt.Int J Pharm 1995;121:19-26.
[26]Kitamori N,Iga K.Dissolution of nonspherical powders. J Pharm Sci 1978;67:1674-1676.
[27]Lu ATK,Frisella ME,Johnson KC.Dissolution modeling: factors affecting the dissolution rates of polydisperse powders.Pharm Res 1993;10:1308-1314.
[28]Nu´n~ez C,Vin~als J,Roca A,et al.A general shrinking-particle model for the chemical dissolution of crystalline forms. Hydrometallurgy 1994;36:1-17.
[29]Ma Z,Merkus HG,De Smet J,et al.New developments in particle characterization by laser diffraction:size and shape.Powder Technol 2000;111:66-78.
[30]Hickey AJ,Ganderton D.Size reduction and classi fi cation. In:Hickey JA,Ganderton D,editors.Pharmaceutical process engineering.New York:Marcel Dekker;2001.p.174-197.
[31]Boldyrev VV.Mechanochemical modi fi cation and synthesis of drugs.J Mater Sci 2004;39:5117-5120.
[32]Hersey JA,Krycer I.Biopharmaceutical implications of technological change.Int J Pharm Tech Prod Manuf 1980;1:18-21.
[33]Hutten-Raunch R.Modi fi cation of starting materials to improve tablet properties.Pharm Ind 1983;45:435-440.
[34]Yu L.Amorphous pharmaceutical solids:preparation, characterization and stabilization.Adv Drug Deliv Rev 2001;48:27-42.
[35]Shakhtshneider TP.Phase transformations and stabilization of metastable states of molecular crystals under mechanical activation.Solid State Ionics 1997;101-103:851-856.
[36]Dudognon E,Willart JF,Caron V,et al.Formation of budesonide/α-lactose glass solutions by ball-milling.Solid State Commun 2006;138:68-71.
[37]Kayaert P,Van Den Mooter G.Is the amorphous fraction of a dried nanosuspension caused by milling or by drying?A case study with naproxen and cinnarizine.Eur J Pharm Biopharm 2012;81:650-656.
[38]Karmwar P,Graeser K,Gordon KC,et al.Effect of different preparation methods on the dissolution behaviour of amorphous indomethacin.Eur J Pharm Biopharm 2012;80:459-464.
[39]Guinot S,Leveiller F.The use of mtdsc to assess the amorphous phase content of a micronised drug substance. Int J Pharm 1999;192:63-75.
[40]Elamin AA,Ahlneck C,Alderborn G,et al.Increased metastable solubility of milled griseofulvin,depending on the formation of a disordered surface-structure.Int J Pharm 1994;111:159-170.
[41]Weinek¨otter R,Gericke H.Mixing and degregation processes.In:Weinekotter R,Gericke H,editors.Mixing of solids.Dordrecht:Kluwer Academic Publishers;2000. p.7-14.
[42]Otte A,Carvajal MT.Assessment of milling-induced disorder of two pharmaceutical compounds.J Pharm Sci 2011;100:1793-1804.
[43]Van Eerdenbrugh B,Van Den Mooter G,Augustijns P.Topdown production of drug nanocrystals:nanosuspension stabilization,miniaturization and transformation into solid products.Int J Pharm 2008;364:64-75.
[44]Bahl D,Bogner RH.Amorphization of indomethacin by cogrinding with neusilin us2:amorphization kinetics, physical stability and mechanism.Pharm Res 2006;23:2317-2325.
[45]Choi JY,Park CH,Lee J.Effect of polymer molecular weight on nanocomminution of poorly soluble drug.Drug Deliv 2008;15:347-353.
[46]Lee J,Choi JY,Park CH.Characteristics of polymers enabling nano-comminution of water-insoluble drugs.Int J Pharm 2008;355:328-336.
[47]Lee J,Lee SJ,Choi JY,et al.Amphiphilic amino acid copolymers as stabilizers for the preparation of nanocrystal dispersion.Eur J Pharm Sci 2005;24:441-449.
[48]Choi JY,Yoo JY,Kwak HS,et al.Role of polymeric stabilizers for drug nanocrystal dispersions.Curr Appl Phys 2005;5:472-474.
[49]Bhakay A,Merwade M,Bilgili E,et al.Novel aspects of wet milling for the production of microsuspensions and nanosuspensions of poorly water-soluble drugs.Drug Dev Ind Pharm 2011;37:963-976.
[50]Ghosh I,Bose S,Vippagunta R,et al.Nanosuspension for improving the bioavailability of a poorly soluble drug and screening of stabilizing agents to inhibit crystal growth.Int J Pharm 2011;409:260-268.
[51]Van Eerdenbrugh B,Vermant J,Martens JA,et al.A screening study of surface stabilization during the production of drug nanocrystals.J Pharm Sci 2009;98:2091-2103.
[52]Saleem IY,Smyth HDC.Micronization of a soft material:airjet and micro-ball milling.AAPS PharmSciTech 2010;11:1642-1649.
[53]Shariare MH,Blagden N,Matas MD,et al.In fl uence of solvent on the morphology and subsequent comminution of ibuprofen crystals by air jet milling.J Pharm Sci 2012;101:1108-1119.
[54]Shariare MH,De Matas M,York P.Effect of crystallisation conditions and feedstock morphology on the aerosolization performance of micronised salbutamol sulphate.Int J Pharm 2011;415:62-72.
[55]Brodka-Pfeiffer K,Hausler HP,Grass P,et al.Air jet milling with homogeneous premixes of fenoterol hydrobromide and glucose for the application in dry powder inhalers. Pharm Ind 2005;67:713-719.
[56]Jain RA,Brito L,Straub JA,et al.Effect of powder processing on performance of feno fi brate formulations.Eur J Pharm Biopharm 2008;69:727-734.
[57]Vogt M,Vertzoni M,Kunath K,et al.Cogrinding enhances the oral bioavailability of emd 57033,a poorly water soluble drug,in dogs.Eur J Pharm Biopharm 2008;68:338-345.
[58]Han X,Ghoroi C,To D,et al.Simultaneous micronization and surface modi fi cation for improvement of fl ow and dissolution of drug particles.Int J Pharm 2011;415:185-195.
[59]Muller RH,Peters K,Becker R,et al.Nanosuspensions-a novel formulation for the iv administration of poorly soluble drugs.In:Proceedings of the 1st world meeting of the international association for pharmaceutical technology,Budapest;1995.p.491-492.
[60]Hoyer H,Schlocker W,Greindl M,et al.Preparation and evaluation of thiomer nanoparticles via high pressure homogenization.J Microencapsul 2010;27:487-495.
[61]Colombo I,Grassi G,Grassi M.Drug mechanochemical activation.J Pharm Sci 2009;98:3961-3986.
[62]Suryanarayana C.Mechanical alloying and milling.Prog Mater Sci 2001;46:1-184.
[63]Lantz RJ.Size reduction.In:Lieberman HA,Lachman L, Schwartz JB,editors.Pharmaceutical dosage forms:Tablets. New York:Marcel Dekker,Inc.;1990.p.107-200.
[64]Yamamoto K,Nakano M,Arita T,et al.Dissolution rate and bioavailability of griseofulvin from a ground mixture with microcrystalline cellulose.J Pharmacokinet Biop 1974;2:487-493.
[65]Yamamoto K,Nakano M,Arita T.Dissolution behavior and bioavailability of phenytoin from a ground mixture with microcrystalline cellulose.J Pharm Sci 1976;65:1484-1488.
[66]Mallick S,Pattnaik S,Swain K,et al.Physicochemical characterization of interaction of ibuprofen by solid-state milling with aluminum hydroxide.Drug Dev Ind Pharm 2008;34:726-734.
[67]Balani PN,Ng WK,Tan RB,et al.In fl uence of excipients in comilling on mitigating milling-induced amorphization or structural disorder of crystalline pharmaceutical actives. J Pharm Sci 2010;99:2462-2474.
[68]L¨obmann K,Strachan C,Grohganz H,et al.Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions.Eur J Pharm Biopharm 2012;81:159-169.
[69]Tozuka Y,Imono M,Uchiyama H,et al.A novel application of α-glucosyl hesperidin for nanoparticle formation of active pharmaceutical ingredients by dry grinding.Eur J Pharm Biopharm 2011;79:559-565.
[70]Mu¨ller RH,Jacobs C,Kayser O.Nanosuspensions as particulate drug formulations in therapy:rationale for development and what we can expect for the future.Adv Drug Deliv Rev 2001;47:3-19.
[71]Peltonen L,Hirvonen J.Pharmaceutical nanocrystals by nanomilling:critical process parameters,particle fracturing and stabilization methods.J Pharm Pharmacol 2010;62:1569-1579.
[72]Gulsun T,Gursoy RN,Oner L.Design and characterization of nanocrystal formulations containing ezetimibe.Chem Pharm Bull 2011;59:41-45.
[73]Sievens-Figueroa L,Bhakay A,Jerez-Rozo JI,et al. Preparation and characterization of hydroxypropyl methyl cellulose fi lms containing stable BCS class ii drug nanoparticles for pharmaceutical applications.Int J Pharm 2012;423:496-508.
[74]Kayaert P,Annˊe M,Van Den Mooter G.Bead layering as a process to stabilize nanosuspensions:in fl uence of drug hydrophobicity on nanocrystal reagglomeration following in-vitro release from sugar beads.J Pharm Pharmacol 2011;63:1446-1453.
[75]Kesisoglou F,Panmai S,Wu Y.Nanosizing-oral formulation development and biopharmaceutical evaluation.Adv Drug Deliv Rev 2007;59:631-644.
[76]Liversidge GG,Cundy KC.Particle-size reduction for improvement of oral bioavailability of hydrophobic drugs.1. Absolute oral bioavailability of nanocrystalline danazol in beagle dogs.Int J Pharm 1995;125:91-97.
[77]Merisko-Liversidge E,Liversidge GG,Cooper ER.Nanosizing: a formulation approach for poorly-water-soluble compounds.Eur J Pharm Sci 2003;18:113-120.
[78]Patravale VB,Date AA,Kulkarni RM.Nanosuspensions:a promising drug delivery strategy.J Pharm Pharmacol 2004;56:827-840.
[79]Verhoff FH,Snow RA,Pace GW.Media milling.US Patent 6604698.2003.
[80]Moschwitzer J,Muller RH.New method for the effective production of ultra fi ne drug nanocrystals.J Nanosci Technol 2006;6:3145-3153.
[81]Van Eerdenbrugh B,Froyen L,Martens JA,et al. Characterization of physico-chemical properties and pharmaceutical performance of sucrose co-freeze-dried solid nanoparticulate powders of the anti-hiv agent loviride prepared by media milling.Int J Pharm 2007;338:198-206.
[82]Park CH,Youn HR,Lee J,et al.Improved ef fi cacy of appetite suppression by lipoic acid particles prepared by nanocomminution.Drug Dev Ind Pharm 2009;35:1305-1311.
[83]Ghosh I,Michniak-Kohn B.Design and characterization of submicron formulation for a poorly soluble drug:the effect of vitamin e tpgs and other solubilizers on skin permeability enhancement.Int J Pharm 2012;434:90-98.
[84]Niwa T,Miura S,Danjo K.Design of dry nanosuspension with highly spontaneous dispersible characteristics to develop solubilized formulation for poorly water-soluble drugs.Pharm Res 2011;28:2339-2349.
[85]Nekkanti V,Pillai R,Venkateshwarlu V,et al.Development and characterization of solid oral dosage form incorporating candesartan nanoparticles solid oral dosage form incorporating drug nanoparticles.Pharm Dev Technol 2009;14:290-298.
[86]Laaksonen T,Liu P,Rahikkala A,et al.Intact nanoparticulate indomethacin in fast-dissolving carrier particles by combined wet milling and aerosol fl ow reactor methods.Pharm Res 2011;28:2403-2411.
[87]Tanaka Y,Inkyo M,Yumoto R,et al.Nanoparticulation of probucol,a poorly water-soluble drug,using a novel wetmilling process to improve in vitro dissolution and in vivo oral absorption.Drug Dev Ind Pharm 2012;38:1015-1023.
[88]Chan HK,Kwok PCL.Production methods for nanodrug particles using the bottom-up approach.Adv Drug Deliv Rev 2011;63:406-416.
[89]Shegokar R,Mu¨ller RH.Nanocrystals:industrially feasible multifunctional formulation technology for poorly soluble actives.Int J Pharm 2010;399:129-139.
[90]Muller RH,Becker R,Kruss B,et al.Pharmaceutical nanosuspensions for medicament administration as system of increased saturation solubility and rate of solution.US Patent No.5858410.1998.
[91]Mohr KH.High-pressure homogenization.Part i.Liquidliquid dispersion in turbulence fi elds of high energy density. J Food Eng 1987;6:177-186.
[92]Pandolfe WD.Effect of dispersed and continuous phase viscosity on droplet size of emulsions generated by homogenization.J Disper Sci Technol 1981;2:459-474.
[93]Jahnke S.The theory of high-pressure homogenization.In: Muller RH,Benita S,Bohm B,editors.Emulsions and nanosuspensions for the formulation of poorly soluble drugs.Stuttgart:Medpharm Scienti fi c;1998.p.177-200.
[94]Mu¨ller RH,Peters K.Nanosuspensions for the formulation of poorly soluble drugs.I.Preparation by a size-reduction technique.Int J Pharm 1998;160:229-237.
[95]Schwarz C,Mehnert W,Muller RH.In fl uence of production parameters of solid lipid nanoparticles(SLN)on the suitability for intravenous injection.Eur J Pharm Biopharm 1994;40.
[96]Radtke M.Nanopure pure drug nanoparticles for the formulation of poorly soluble drugs.New Drugs 2001;3:62-68.
[97]Kluge J,Mazzotti M.Co 2-assisted high pressure homogenization:a solvent-free process for polymeric microspheres and drug-polymer composites.Int J Pharm 2012;436:394-402.
[98]Kluge J,Muhrer G,Mazzotti M.High pressure homogenization of pharmaceutical solids.J Supercrit Fluid 2012;66:380-388.
[99]Quan P,Shi K,Piao H,et al.A novel surface modi fi ed nitrendipine nanocrystals with enhancement of bioavailability and stability.Int J Pharm 2012;430:366-371.
[100]Ye J,Schoenung JM.Technical cost modeling for the mechanical milling at cryogenic temperature(cryomilling). Adv Eng Mater 2004;6.656-664+611.
[101]Salazar J,Ghanem A,Mu¨ller RH,et al.Nanocrystals: comparison of the size reduction effectiveness of a novel combinative method with conventional top-down approaches.Eur J Pharm Biopharm 2012;81:82-90.
[102]Sugimoto S,Niwa T,Nakanishi Y,et al.Development of a novel ultra cryo-milling technique for a poorly watersoluble drug using dry ice beads and liquid nitrogen.Int J Pharm 2012;426:162-169.
[103]Yang YT,Chen CT,Yang JC,et al.Spray-dried microparticles containing polymeric micelles encapsulating hematoporphyrin.AAPS J 2010;12:138-146.
[104]Shakhtshneider TP,Boldyrev VV.Mechanochemical synthesis and mechanical activation of drugs.In: Boldyreva E,Boldyrev VV,editors.Reactivity of molecular solids.Chichester,GB:John Wiley&Sons Ltd.;1999. p.271-311.
[105]Sugimoto S,Niwa T,Nakanishi Y,et al.Novel ultra-cryo milling and co-grinding technique in liquid nitrogen to produce dissolution-enhanced nanoparticles for poorly water-soluble drugs.Chem Pharm Bull 2012;60:325-333.
[106]Jayasankar A,Somwangthanaroj A,Shao ZJ,et al.Cocrystal formation during cogrinding and storage is mediated by amorphous phase.Pharm Res 2006;23:2381-2392.
[107]Kogermann K,Veski P,Rantanen J,et al.X-ray powder diffractometry in combination with principal component analysis-a tool for monitoring solid state changes.Eur J Pharm Sci 2011;43:278-289.
[108]Willart JF,Descamps M.Solid state amorphization of pharmaceuticals.Mol Pharm 2008;5:905-920.
[109]Crowley KJ,Zogra fiG.Cryogenic grinding of indomethacin polymorphs and solvates:assessment of amorphous phase formation and amorphous phase physical stability.J Pharm Sci 2002;91:492-507.
[110]Karmwar P,Graeser K,Gordon KC,et al.Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods.Int J Pharm 2011;417:94-100.
[111]Wojnarowska Z,Grzybowska K,Adrjanowicz K,et al.Study of the amorphous glibenclamide drug:analysis of the molecular dynamics of quenched and cryomilled material. Mol Pharm 2010;7:1692-1707.
[112]Chieng N,Rades T,Saville D.Formation and physical stability of the amorphous phase of ranitidine hydrochloride polymorphs prepared by cryo-milling.Eur J Pharm Biopharm 2008;68:771-780.
[113]Adrjanowicz K,Kaminski K,Grzybowska K,et al.Effect of cryogrinding on chemical stability of the sparingly watersoluble drug furosemide.Pharm Res 2011;28:3220-3236.
[114]Niwa T,Nakanishi Y,Danjo K.One-step preparation of pharmaceutical nanocrystals using ultra cryo-milling technique in liquid nitrogen.Eur J Pharm Sci 2010;41:78-85.
[115]Feng T,Pinal R,Carvajal MT.Process induced disorder in crystalline materials:differentiating defective crystals from the amorphous form of griseofulvin.J Pharm Sci 2008;97:3207-3221.
[116]Ma Z,Merkus HG,Van Der Veen HG,et al.On-line measurement of particle size and shape using laser diffraction.Part Part Syst Char 2002;18:243-247.
[117]Deriemaeker L,Finsy R.Shape and size determination by laser diffraction:average aspect ratio and size distributions by volume;feasibility of data analysis by neural networks. Part Part Syst Char 2005;22:5-13.
[118]Ma Z,Merkus HG,Scarlett B.Extending laser diffraction for particle shape characterization:technical aspects and application.Powder Technol 2001;118:180-187.
[119]Xu R,Di Guida OA.Comparison of sizing small particles using different technologies.Powder Technol 2003;132:145-153.
[120]Almeida-Prieto S,Blanco-Mˊendez J,Otero-Espinar FJ.Image analysis of the shape of granulated powder grains.J Pharm Sci 2004;93:621-634.
[121]Staniforth J.Particle-size reduction.In:Aulton M,editor. Pharmaceutics:the science of dosage form design.2nd ed. London:Churchill Livingstone;2002.p.166-173.
[122]George M,Ghosh I.Identifying the correlation between drug/stabilizer properties and critical quality attributes (cqas)of nanosuspension formulation prepared by wet media milling technology.Eur J Pharm Sci 2013;48:142-152.
[123]Bilgili E,Afolabi A.A combined microhydrodynamicspolymer adsorption analysis for elucidation of the roles of stabilizers in wet stirred media milling.Int J Pharm 2012;439:193-206.
[124]Ghosh I,Schenck D,Bose S,et al.Optimization of formulation and process parameters for the production of nanosuspension by wet media milling technique:effect of vitamin e tpgs and nanocrystal particle size on oral absorption.Eur J Pharm Sci 2012;47:718-728.
[125]Anhalt K,Geissler S,Harms M,et al.Development of a new method to assess nanocrystal dissolution based on light scattering.Pharm Res 2012;29:2887-2901.
[126]De Smet L,Colin P,Ceelen W,et al.Development of a nanocrystalline paclitaxel formulation for hipec treatment. Pharm Res 2012;29:2398-2406.
[127]Bose S,Schenck D,Ghosh I,et al.Application of spray granulation for conversion of a nanosuspension into a dry powder form.Eur J Pharm Sci 2012;47:35-43.
[128]Niwa T,Miura S,Danjo K.Universal wet-milling technique to prepare oral nanosuspension focused on discovery and preclinical animal studies-development of particle design method.Int J Pharm 2011;405:218-227.
[129]Cerdeira AM,Mazzotti M,Gander B.Miconazole nanosuspensions:in fl uence of formulation variables on particle size reduction and physical stability.Int J Pharm 2010;396:210-218.
[130]Jinno J,Kamada N,Miyake M,et al.In vitro-in vivo correlation for wet-milled tablet of poorly water-soluble cilostazol.J Control Release 2008;130:29-37.
[131]Merisko-Liversidge E,Mcgurk SL,Liversidge GG.Insulin nanoparticles:a novel formulation approach for poorly water soluble zn-insulin.Pharm Res 2004;21:1545-1553.
[132]Na GC,Stevens Jr HJ,Yuan BO,et al.Physical stability of ethyl diatrizoate nanocrystalline suspension in steam sterilization.Pharm Res 1999;16:569-574.
[133]Sangwai M,Vavia P.Amorphous ternary cyclodextrin nanocomposites of telmisartan for oral drug delivery: improved solubility and reduced pharmacokinetic variability.Int J Pharm 2013;453:423-432.
[134]Scalia S,Franceschinis E,Bertelli D,et al.Comparative evaluation of the effect of permeation enhancers,lipid nanoparticles and colloidal silica on in vivo human skin penetration of quercetin.Skin Pharmacol Phys 2012;26:57-67.
[135]Talekar M,Kendall J,Denny W,et al.Development and evaluation of pik75 nanosuspension,a phosphatidylinositol-3-kinase inhibitor.Eur J Pharm Sci 2012;47:824-833.
[136]Rachmawati H,Shaal LA,Mu¨ller RH,et al.Development of curcumin nanocrystal:physical aspects.J Pharm Sci 2013;102:204-214.
[137]Muller RH,Harden D,Keck CM.Development of industrially feasible concentrated 30%and 40%nanoemulsions for intravenous drug delivery.Drug Dev Ind Pharm 2012;38:420-430.
[138]Chu T,Zhang Q,Li H,et al.Development of intravenous lipid emulsion of tanshinone iia and evaluation of its antihepatoma activity in vitro.Int J Pharm 2012;424:76-88.
[139]Sun W,Tian W,Zhang Y,et al.Effect of novel stabilizerscationic polymers on the particle size and physical stability of poorly soluble drug nanocrystals.Nanotechnol Biol Med 2012;8:460-467.
[140]Wang S,Chen T,Chen R,et al.Emodin loaded solid lipid nanoparticles:preparation,characterization and antitumor activity studies.Int J Pharm 2012;430:238-246.
[141]Xu LM,Zhang QX,Zhou Y,et al.Engineering drug ultra fi ne particles of beclomethasone dipropionate for dry powder inhalation.Int J Pharm 2012;436:1-9.
[142]Zhang J,Lv H,Jiang K,et al.Enhanced bioavailability after oral and pulmonary administration of baicalein nanocrystal.Int J Pharm 2011;420:180-188.
[143]Jeon SO,Hwang HJ,Oh DH,et al.Enhanced percutaneous delivery of recombinant human epidermal growth factor employing nano-liposome system.J Microencapsul 2012;29:234-241.
[144]Anwar M,Warsi MH,Mallick N,et al.Enhanced bioavailability of nano-sized chitosan-atorvastatin conjugate after oral administration to rats.Eur J Pharm Sci 2011;44:241-249.
[145]Wang L,Hao Y,Liu N,et al.Enhance the dissolution rate and oral bioavailability of pranlukast by preparing nanosuspensions with high-pressure homogenizing method.Drug Dev Ind Pharm 2012;38:1381-1389.
[146]Kakran M,Shegokar R,Sahoo NG,et al.Fabrication of quercetin nanocrystals:comparison of different methods. Eur J Pharm Biopharm 2012;80:113-121.
[147]He W,Tan Y,Tian Z,et al.Food protein-stabilized nanoemulsions as potential delivery systems for poorly water-soluble drugs:preparation,in vitro characterization, and pharmacokinetics in rats.Int J Nanomed 2011;6:521-533.
[148]Kim DM,Hyun SS,Yun P,et al.Identi fi cation of an emulsi fi er and conditions for preparing stable nanoemulsions containing the antioxidant astaxanthin.Int J Cosmet Sci 2012;34:64-73.
[149]Gonzalez-Mira E,Nikoliˊc S,Calpena AC,et al.Improved and safe transcorneal delivery of fl urbiprofen by nlc and nlcbased hydrogels.J Pharm Sci 2012;101:707-725.
[150]Wang Y,Ma Y,Zheng Y,et al.In vitro and in vivo anticancer activity of a novel puerarin nanosuspension against colon cancer,with high ef fi cacy and low toxicity.Int J Pharm 2013;441:728-735.
[151]Yao L,Zhao X,Li Q,et al.In vitro and in vivo evaluation of camptothecin nanosuspension:a novel formulation with high antitumor ef fi cacy and low toxicity.Int J Pharm 2012;423:586-588.
[152]Piao H,Ouyang M,Xia D,et al.In vitro-in vivo study of coq10-loaded lipid nanoparticles in comparison with nanocrystals.Int J Pharm 2011;419:255-259.
[153]Kakran M,Shegokar R,Sahoo NG,et al.Long-term stability of quercetin nanocrystals prepared by different methods. J Pharm Pharmacol 2012b;64:1394-1402.
[154]Mitri K,Shegokar R,Gohla S,et al.Lutein nanocrystals as antioxidant formulation for oral and dermal delivery.Int J Pharm 2011;420:141-146.
[155]Schwarz JC,Weixelbaum A,Pagitsch E,et al.Nanocarriers for dermal drug delivery:in fl uence of preparation method, carrier type and rheological properties.Int J Pharm 2012;437:83-88.
[156]Lai F,Pini E,Angioni G,et al.Nanocrystals as tool to improve piroxicam dissolution rate in novel orally disintegrating tablets.Eur J Pharm Biopharm 2011;79:552-558.
[157]Duret C,Wauthoz N,Sebti T,et al.New inhalationoptimized itraconazole nanoparticle-based dry powders for the treatment of invasive pulmonary aspergillosis.Int J Nanomed 2012;7:5475-5489.
[158]Botker JP,Karmwar P,Strachan CJ,et al.Assessment of crystalline disorder in cryo-milled samples of indomethacin using atomic pair-wise distribution functions.Int J Pharm 2011;417:112-119.
[159]Garmise RJ,Mar K,Crowder TM,et al.Formulation of a dry powder in fl uenza vaccine for nasal delivery.AAPS Pharm Sci Tech 2006;7:E1-7.
Zhi Hui Loha,Asim Kumar Samantab,Paul Wan Sia Henga,*
aGEANUS Pharmaceutical Processing Research Laboratory,Department of Pharmacy,National University of Singapore,18 Science Drive 4,Singapore 117543,SingaporebJanssen India,Department of PDMS-SMMD-AD,Johnson&Johnson Ltd.,Higi House,Opp Ralli Wolf,LBS Marg, Mulund(W),Mumbai 400080,India
*Corresponding author.GEANUS Pharmaceutical Processing Research Laboratory,Department of Pharmacy,National University of Singapore,18 Science Drive 4,Singapore 117543,Singapore.Tel.:+65 6516 2930;fax:+65 6779 1554.
E-mail address:phapaulh@nus.edu.sg(P.W.Sia Heng).
Peer review under responsibility of Shenyang Pharmaceutical University.
http://dx.doi.org/10.1016/j.ajps.2014.12.006
1818-0876/©2015 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.This is an open access article under the CC BY-NC-ND license(http://creativecommons.org/licenses/by-nc-nd/4.0/).
 Asian Journal of Pharmacentical Sciences2015年4期
Asian Journal of Pharmacentical Sciences2015年4期
- Asian Journal of Pharmacentical Sciences的其它文章
- GUIDE FOR AUTHORS
- Effect of addition of inulin and fenugreek on the survival of microencapsulated Enterococcus durans 39C in alginate-psyllium polymeric blends in simulated digestive system and yogurt
- Zingiber cassumunar blended patches for skin application:Formulation,physicochemical properties,and in vitro studies
- Taste masking of cipro fl oxacin by ion-exchange resin and sustain release at gastric-intestinal through interpenetrating polymer network
- Design and development of novel bioadhesive niosomal formulation for the transcorneal delivery of anti-infective agent:In-vitro and ex-vivo investigations
- Effect of formulation variables on in vitro release of a water-soluble drug from chitosan-sodium alginate matrix tablets