
腓骨肌萎缩症—家系报道
2015-05-08 07:22:42黄丽敏江新梅高凤娟
中国实验诊断学 2015年9期
黄丽敏,江新梅,常 颖,高凤娟
(1.吉林大学中日联谊医院,吉林长春130033;2.吉林大学第一医院;3.桦旬市新华社区卫生服务中心)
腓骨肌萎缩症—家系报道
黄丽敏1,江新梅2*,常 颖1,高凤娟3
(1.吉林大学中日联谊医院,吉林长春130033;2.吉林大学第一医院;3.桦旬市新华社区卫生服务中心)
腓骨肌萎缩症(CMT)占遗传性周围神经病的80%-90%,临床特征为[1]双足内侧肌和腓骨肌进行性无力和萎缩,累及双上肢时呈爪形手,可伴有弓形足及垂足,双下肢腱反射尤其是踝反射减弱或消失,部分病人可伴有轻到中度感觉减退。现将诊断为CMT一家系进行报道,总结临床特点。
1 临床资料
先证者,女,47岁,双下肢活动不灵近13年,双上肢活动不灵近6年就诊。34岁时发现双下肢抬举无力,劳累后行走费力,缓慢进展,6年前出现步行困难及双手精细运动差,现需拄拐行走。查体见双侧大腿以下肌肉萎缩,呈鹤腿,有弓形足,四肢腱反射减弱,浅感觉正常,双下肢深感觉消失。肌电图结果示双上肢及双下肢神经源性损害,下肢受累程度重于上肢,感觉、运动神经均受累。应用等位基因特异性PCR-双酶切方法[1]进行PMP22基因重复突变检测结果为阴性。
家系调查:该家系4代共45人,其中男性18人,女性27人,发病12人,全部为女性。本家系第1代平均发病年龄为51.5岁,第2代为30.8岁,第3代为17.3岁。Ⅱ2因脑梗死去世,去世前有双下肢无力及肌萎缩。所有发病者均存在腱反射异常,首发症状为双下肢无力,表现为双下肢容易疲劳,走路时容易摔倒,逐渐出现比同龄人跑跳差或难以跑跳,缓慢进展出现上楼困难。此家系中病情最重者为Ⅰ1与Ⅰ2,双下肢呈“倒置酒瓶样”改变及弓形足,需用轮椅维持日常生活。病情次重者为Ⅱ8与Ⅱ9,现用手拐才能行走,病情较轻者为Ⅱ5、Ⅱ6与Ⅱ10,现不能跑跳,无肌萎缩。病情最轻者为Ⅲ5、Ⅲ13、Ⅲ18与Ⅲ20,现双下肢易疲劳且比同龄人跑跳差,无肌萎缩。所有发病者在整个病程中无主观感觉障碍的叙述,但查体发现深感觉减弱或消失。Ⅲ4、Ⅲ15及Ⅲ23(未发病者)无肢体活动不灵、无肌萎缩且四肢腱反射正常,发现深感觉减弱。

图1 家系图谱
2 讨论
CMT是遗传性周围神经病中一组最常见的疾病,多在儿童或青少年期起病,CMT1型12岁左右,CMT2型25岁左右。据陈嵘等[2]统计79.0%的CMT患者先累及下肢,且大部分为对称受累,少数患者可以出现受累程度不对称[3],表现为一侧先起病,一侧肌萎缩较另一侧明显或弓形足不对称。此家系起病时受累部位均为下肢,且对称受累。CMT最突出的临床特征是对称性双下肢远端肌无力和(或)肌萎缩,典型者呈“鹤腿”或“倒置酒瓶样”改变[4],部分病人可有双上肢受累呈爪形手,一般下肢重于上肢。此家系所有发病者均有肌无力症状,病情严重者伴有足内侧肌和腓骨肌萎缩及爪形手,下肢受累较上肢重。所有发病者均有腱反射减弱或者消失,且腱反射减弱越明显,肌萎缩越严重,与任雪芳等[5]报道相似。陈嵘等统计[2]国内CMT感觉障碍的发生率为39.6%。此家系所有发病者查体均发现深感觉减弱或者消失,3名未发病者(Ⅲ4、Ⅲ15、Ⅲ23)发现深感觉减弱,但拒绝行肌电图等任何检查,无法诊断为CMT。CMT的遗传方式多数为常染色体显性,少数为常染色体隐性及X连锁遗传[6]。本病在此家系中连续三代都发病,为显性遗传,且本家系中尚无明确的男性成员发病,发病者全部为女性,因此遗传方式以常染色体显性遗传可能性最大。此家系每代发病年龄提前,存在遗传早现现象[7]。此家系排除因PMP22基因重复突变导致的CMT1A型。
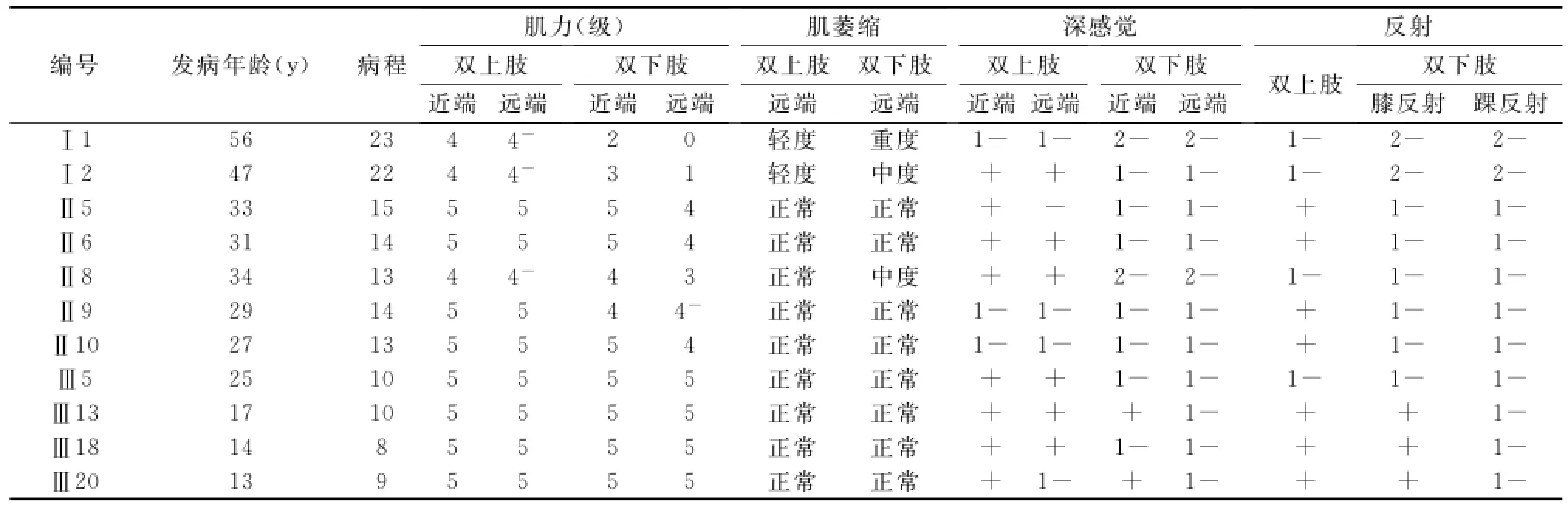
表2 11名发病者临床资料
[1]汪仁斌,严 莉,段晓慧,等.周围髓鞘蛋白22基因重复突变致夏科-马里-图斯病1A亚型的临床和神经电生理特征[J].脑与神经疾病杂志,2012,20(2):92.
[2]陈 嵘,梁秀龄.中国腓骨肌萎缩症的临床特点[J].临床神经病学杂志,1996,9(6):345.
[3]陈 嵘,梁秀龄.腓骨肌萎缩症的临床表现和分型及遗传学[J].中山医科大学学报,1997,18(3):213.
[4]邹 艺,刘 英,李素荣,等.腓骨肌萎缩症的临床及神经电生理特点分析[J].临床神经电生理学杂志,2008,17(5):268.
[5]任雪芳,吴保仁,粟秀初.腓骨肌萎缩症29例[J].第四军医大学学报,1986,7(3):247.
[6]周向平,张 成.Charcot-Marie-Tooth的分子机制[J].国外医学遗传学分册,2000,23(6):297.
[7]张家良,常建军.腓骨肌萎缩症1型临床与电生理分析[J].中国康复,2005,20(1):39.
2014-08-28)
1007-4287(2015)09-1592-02
*通讯作者
猜你喜欢
吉林大学学报(地球科学版)(2023年5期)2023-11-29 02:02:06
吉林大学学报(理学版)(2021年5期)2021-09-22 05:53:02
吉林大学学报(理学版)(2021年2期)2021-03-25 01:15:26
防爆电机(2020年4期)2020-12-14 03:11:16
吉林大学学报(理学版)(2020年6期)2020-11-26 01:05:12
高原山地气象研究(2016年2期)2016-11-10 06:06:36
中西医结合心脑血管病杂志(2016年20期)2016-03-01 04:20:48
云南畜牧兽医(2015年4期)2015-02-28 21:26:11
西安交通大学学报(医学版)(2015年2期)2015-02-28 17:59:20
当代畜禽养殖业(2014年10期)2014-02-27 07:59:48
