
利用T7核酸外切酶和硫代磷酸化修饰引物克隆长片段基因的研究
2015-03-24 07:52张玉祥
解放军医药杂志 2015年8期
兰 泓,张玉祥
·论著·
利用T7核酸外切酶和硫代磷酸化修饰引物克隆长片段基因的研究
兰 泓,张玉祥
目的 采用不依赖连接反应的克隆法,利用T7核酸外切酶和硫代磷酸化修饰引物克隆Notch2长片段基因。方法 将难以扩增的Notch2 cDNA的编码序列(7416 bp)人为分成3段,引物设计时对此3个片段和载体骨架的引物进行碱基硫代磷酸化修饰,用这些引物扩增Notch2的3个片段和载体骨架。然后用T7核酸外切酶分别处理PCR产物,产生4个具有3′互补突出末端的片段,并将此4个末端突出的片段退火复性,完成Notch2基因克隆。结果 琼脂糖凝胶电泳结果显示Notch2 3个片段和载体骨架的PCR产物大小与预期大小相符,经退火复性获得的克隆通过PCR、酶切和测序进行克隆鉴定,确定Notch2编码序列已经插入到pcDNA3.0-3*Flag载体中。结论 利用T7核酸外切酶和引物的碱基磷酸化修饰进行的不依赖连接反应的克隆方法能够用于长片段基因的克隆。
Notch2基因;长片段;不依赖连接反应;T7核酸外切酶;引物硫代磷酸化修饰
Notch家族蛋白在细胞分化、发育过程中起重要作用,与肿瘤的发生、发展有关。哺乳动物Notch家族有4个Notch受体(Notch1~4)。虽然这4个Notch蛋白的基本结构相同,但是它们也有各自不同的特点和功能[1-2]。为了进一步研究Notch蛋白的功能,克隆Notch基因是必要的。Notch2基因cDNA全长11 474 bp,其编码序列7416 bp。一般PCR通常能扩增2~3 kb以内的短片段,对于5 kb以上的长片段很难有效扩增,而且7 kb的长片段与载体的连接效率也比较低,所以采用经典的限制性内切酶方法克隆Notch2基因是比较困难的。本研究利用T7核酸外切酶和引物硫代磷酸化修饰来完成Notch2基因的克隆。
1 材料与方法
1.1 材料
1.1.1 主要试剂:PrimeSTAR GXL DNA polymerase及其缓冲液和dNTP为Takara产品,购自宝生物工程(大连)有限公司;TransStart FastPfu DNA聚合酶和TransT1感受态细胞购自北京全式金生物技术有限公司;DNA液体回收和胶回收试剂盒为MN产品,购自北方仪涛商贸有限公司;T7核酸外切酶为NEB产品,购自北京友谊中联生物科技有限公司;质粒小提试剂盒为Omega产品,购自普京康利生物科技有限公司;10×退火缓冲液自行配制(100 mmol/L Tris-HCl, pH 8.0; 1 mmol/L NaCl; 10 mmol/L EDTA);LB细菌培养液自行配制(胰蛋白胨10 g/L、酵母抽提物5 g/L、NaCl 10 g/L)。pcDNA3.0-3*flag质粒由本实验室自行构建。
1.1.2 引物:本实验所有引物(表1)均由生工生物工程(上海)股份有限公司合成。

表1 引物及其序列
注:*表示经硫代磷酸化修饰
1.2 方法
1.2.1 扩增载体骨架和长片段基因Notch2:以线性化载体pcDNA3.0-3*flag为模板,在TransStart FastPfu DNA聚合酶的作用下扩增载体骨架。以BxPC3细胞系的cDNA为模板,TransStart FastPfu DNA聚合酶的作用下扩增Notch2片段A和片段B,在PrimeSTAR GXL DNA聚合酶的作用下扩增Notch2片段C。相应的扩增引物见表1。PCR条件:95℃预变性2 min;95℃变性20 s,55℃退火20 s,72℃延伸2~3 min(Notch2片段A和片段B延伸2 min、Notch2片段C和载体骨架延伸3 min),30个循环;72℃延伸5 min。
1.2.2 T7核酸外切酶处理、退火复性:分别取已纯化的0.5 μg Notch2 3个片段(A、B、C)和1.0 μg载体骨架,T7核酸外切酶在25℃酶切5 min后,用PCR液体回收试剂盒纯化酶切产物。Notch2的3个已纯化酶切产物按等摩尔比混合,加上0.5倍摩尔数的已纯化载体骨架酶切产物进行退火复性。退火程序为:75℃ 10 min后,每隔90 s降1℃,直至降到25℃。
1.2.3 退火产物的克隆及鉴定:退火产物按照常规细菌转化方法转化TransT1感受态细胞,以已纯化的载体骨架酶切产物转化为阴性对照。挑取2个单克隆按常规方法提取质粒,通过BamHI和NdeI酶切和PCR鉴定出阳性克隆。质粒PCR用pcDNA3.0-3*Flag载体上的通用引物T7和SP6扩增。酶切产物和PCR产物都通过琼脂糖凝胶电泳以观察其条带位置。最后,将酶切和PCR鉴定均为阳性的质粒送生工生物工程(上海)股份有限公司进行测序。测序时使用载体上的测序通用引物T7和SP6分别进行正向和反向测序。
2 结果
2.1 3个Notch2片段和载体骨架PCR扩增 将难以扩增的长片段基因Notch2的cDNA编码序列分成3个片段(A、B、C),其片段大小分别为1877、2504、3095 bp。PCR扩增这3个片段以及pcDNA3.0-3*Flag载体骨架(5432 bp)。琼脂糖凝胶电泳结果显示片段A、B、C和载体骨架4个样品分别在2、2~3、3 、5~6 kb处有明显特异条带,且与预期大小均相符。PCR结果见图1。
2.2 质粒酶切鉴定 挑取2个单克隆,按常规方法提取质粒,用BamHI分别对质粒进行单酶切鉴定,结果显示在10 kb以上出现单酶切条带;用NdeI分别对质粒进行酶切鉴定,结果显示在5~6、0.7~0.8 kb处出现明显条带;用NdeI和BamHI分别对质粒进行双酶切鉴定,经1.1%琼脂糖凝胶电泳可见4条条带,位于5~6、3.0、0.7~0.8 kb处。除了NdeI的酶切结果只显示2条条带,其他酶切(BamHI单酶切和BamHI、NdeI双酶切)条带位置与预期的大小相符,基本可说明所挑取克隆为阳性克隆。质粒酶切鉴定结果见图2。
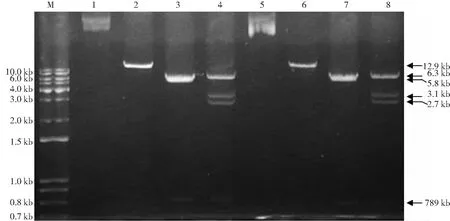
图2 质粒酶切鉴定结果
2.3 质粒PCR 重组质粒PCR扩增产物经0.8%琼脂糖凝胶电泳,可见约8.0 kb处明显特异性条带,与预期大小相符(7570 bp),见图3。
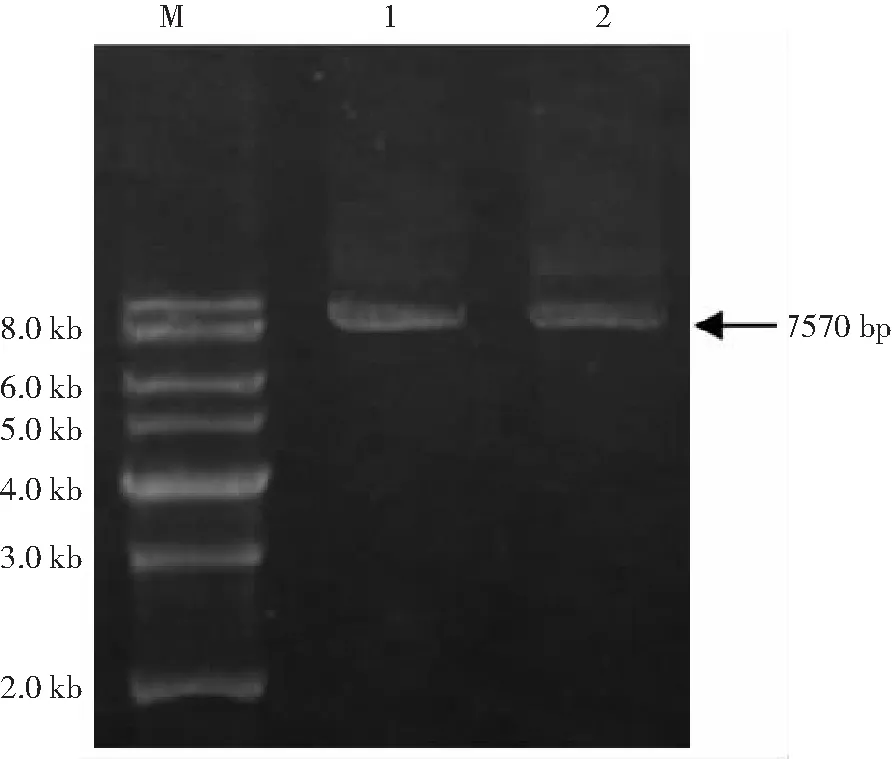
图3 重组质粒的PCR结果
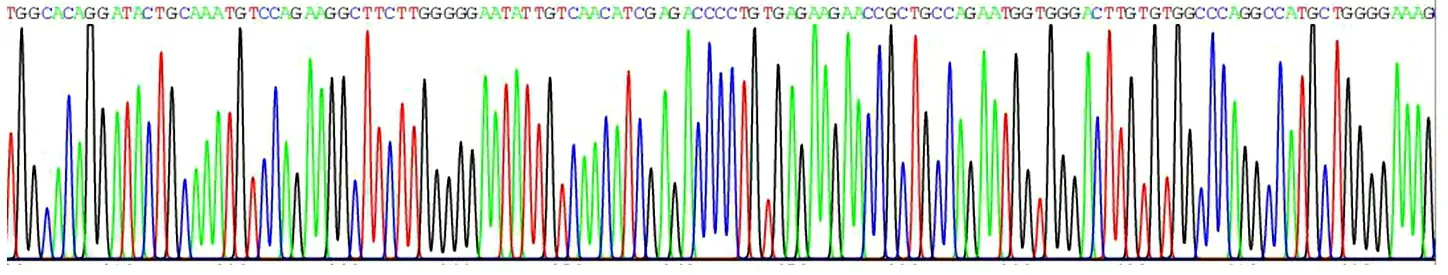
2.4 测序 将2个基本鉴定为阳性的重组质粒用载体上通用引物T7和SP6进行双向测序。测序证实,Notch2编码序列很好地装载入pcDNA3.0-3*Flag载体中,而且通过测序获得的Notch2序列与GeneBank中登录号为NM_024408的Notch2序列一致。两个质粒均测序成功,但图4只显示其中1个质粒的测序结果。
3 讨论
基因克隆技术是分子生物学常用的实验技术。这项技术从20世纪70年代发明以来,经历了许多改进和发展。到目前为止,已经有多项基因克隆技术出现,包括限制性内切酶克隆法、TA克隆、不依赖连接酶的克隆方法[3]、GateWay 技术[4]等。这些技术各有特点,其中的限制性内切酶克隆法最为经典,也最为广泛应用。本实验室曾经试图通过限制性内切酶的方法克隆Notch2基因,但克隆失败,其原因就在于未能直接从cDNA中扩增出Notch2编码序列(7.416 kb)全长。因此,面对这么长的DNA序列的克隆,选择不依赖连接反应的克隆方法(ligation-independent cloning, LIC)。
A
B
LIC是利用具有核酸外切活性的DNA修饰酶分别处理目的片段和载体的PCR产物,产生5′或3′突出互补末端,利用突出的互补末端之间的退火互补进行基因克隆。该方法克服了高背景、低连接效率和受限制性内切酶酶切位点限制等问题,被许多研究者用于质粒构建中[5-6]。最初在1990年Aslanidis和de Jong创建该方法的时候是使用T4 DNA聚合酶处理PCR产物[3],随后其他DNA修饰酶也运用到此方法中,如核酸外切酶III[7]、λ核酸外切酶[8]、尿嘧啶-DNA糖基化酶[9]、USER酶(尿嘧啶特异切除反应酶)[10-11]等。本实验使用具有5′→3′核酸外切活性的T7核酸外切酶处理PCR产物,产生3′突出互补末端,通过突出末端间的简单退火复性,将Notch2基因装载入pcDNA3.0-3*Flag载体中。至于多个DNA片段的无缝克隆,只要用于PCR的引物没有附加其他序列就可以做到。Li和Elledge[12-13]于2007年建立的不依赖序列和连接反应的克隆方法(sequence-and ligation-independent cloning, SLIC)能够做到无缝克隆,但是该方法是通过控制T4 DNA聚合酶的处理时间以及加入dCTP来终止酶切反应。基于碱基硫代磷酸化的克隆方法也是SLIC[14],同样能够做到无缝克隆。Blanusa等[14]在确立该方法时是在碱性溶液中加入碘(乙醇)来切断碱基间的硫代磷酸键。本实验则是通过碱基的硫代磷酸化修饰,以拦截T7核酸外切酶的酶切,从而控制酶切程度。
因为7 kb长的Notch2序列难以直接从cDNA中扩增得到,本实验将Notch2编码序列人为分成易于扩增的3个片段,片段大小分别在2~3 kb。为了使3个Notch2片段之间达到无缝拼接,引物设计时没有添加多余序列,而且在引物中引入碱基的硫代磷酸化修饰。碱基的硫代磷酸化修饰能够阻止T7核酸外切酶对底物的水解[15-17],也就是说,当T7核酸外切酶5′→3′逐个水解底物DNA遇到硫代磷酸化修饰碱基的时候,其水解过程会受到减弱或抑制。Nikiforov等[17]的实验证明4个碱基的硫代磷酸化修饰能够完全阻止此水解过程(室温酶解1 h),但这也增加了实验成本。本实验在适当控制T7核酸外切酶酶解DNA底物的温度和时间(25℃酶解5 min)的前提下,只对1个碱基进行硫代磷酸化修饰,一方面节约了试剂费用,另一方面能较好地控制其酶切程度,获得符合实验条件的3′突出互补末端。
为了验证构建好的质粒的确是Notch2基因的质粒,本研究采用3种方法来鉴定。第1种方法就是选用BamHI和NdeI进行质粒的单酶切和双酶切。Notch2重组质粒中存在3个NdeI酶切位点,分别位于载体片段、Notch2片段A和片段C中,而BamHI酶切位点仅分布在Notch2片段B中。这样通过BamHI和NdeI双酶切酶切产物的大小就可以判断这4个片段是否存在、是否完整及其连接顺序是否正确。图2显示,BamHI单酶切时大于10 kb的位置有一明显的特异条带。NdeI单酶切后在800 bp和6.0 kb处有条带,似乎与预期的酶切后3条条带(789 bp及5.8、6.3 kb)不符。但仔细观察6.0 kb处的条带便可发现,与相邻泳道同一位置的条带相比,这条条带宽度和亮度几乎是相邻条带的两倍,而且该电泳凝胶的浓度为1.1%,此浓度的凝胶极有可能无法分辨5.8 kb和6.3 kb的条带,因此,NdeI单酶切后极有可能有789 bp及5.8、6.3 kb 3个酶切片段。再者,通过BamHI和NdeI双酶切的结果进一步确定Notch2重组质粒经NdeI单酶切酶切产物有789 bp及5.8 、6.3 kb 3个片段。Notch2重组质粒经BamHI和NdeI双酶切,酶切产物理论上应该有4个片段(789 bp及2.7、3.1、6.3 kb),图2的结果与理论上的预期结果相符。第2种方法就是重组质粒PCR。使用载体上的通用引物T7和SP6扩增重组质粒,0.8%琼脂糖凝胶电泳显示8.0 kb左右处有一条明显的特异条带,这也与预期的PCR产物大小(7570 bp)相符。通过这两种方法基本可以确定构建好的重组质粒是目的质粒。第3种方法是对重组质粒测序。本实验使用pcDNA3.0-3*Flag载体的测序引物T7和SP6进行双向测序。测序确定,Notch2编码序列与pcDNA3.0-3*Flag载体很好拼接,而且通过测序获得的Notch2序列与GeneBank中登录号为NM_024408的Notch2序列一致。
这3种方法从多个角度证实了通过本实验方法克隆获得的重组质粒为pcDNA3.0-3*Flag-Notch2目的质粒:第一,从重组质粒PCR结果来看,PCR扩增获得的插入到载体中的Notch2片段大小与Notch2编码序列全长相符;第二,从重组质粒酶切结果来看,Notch2的3个片段和载体骨架均存在该于重组质粒中,而且其前后排列顺序也是正确的;第三,从重组质粒PCR和酶切结果来看,该重组质粒是完整的;第四,从测序结果来看,pcDNA3.0-3*Flag载体和Notch2基因的拼接是无误的,且插入载体的片段确定为Notch2基因。
本研究在引物设计时引入碱基硫代磷酸化修饰,并用T7核酸外切酶处理目的片段和载体的PCR产物,产生3'突出互补末端,突出互补末端间的退火复性而重组DNA,成功构建了pcDNA3.0-3*Flag-Notch2重组质粒。该方法的成功运用为长片段基因的克隆提供了一种思路。
[1] Dell'albani P, Rodolico M, Pellitteri R,etal. Differential patterns of NOTCH1-4 receptor expression are markers of glioma cell differentiation[J].Neuro Oncol, 2014,16(2):206-216.
[2] Pierfelice T J, Schreck K C, Dang L,etal. Notch3 activation promotes invasive glioma formation in a tissue site-specific manner[J].Cancer Res, 2011,71(3):1115-1125.
[3] Aslanidis C, de Jong P J. Ligation-independent cloning of PCR products (LIC-PCR)[J].Nucleic Acids Res, 1990,18(20):6069-6074.
[4] Hartley J L, Temple G F, Brasch M A. DNA Cloning Using In Vitro Site-Specific Recombination[J].Genome Res, 2000,10(11):1788-1795.
[5] De Rybel B, van den Berg W, Lokerse A,etal. A versatile set of ligation-independent cloning vectors for functional studies in plants[J].Plant Physiol, 2011,156(3):1292-1299.
[6] Schmid Burgk J L, Schmidt T, Kaiser V,etal. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes[J].Nat Biotechnol, 2013,31(1):76-81.
[7] Hsiao K. Exonuclease III induced ligase-free directional subcloning of PCR products[J].Nucleic Acids Res, 1993,21(23):5528-5529.
[8] Tseng H. DNA Cloning without Restriction Enzyme and Ligase[J].Bio Techniques, 1999,27(6):1240-1244.
[9] Nisson P E, Rashtchian A, Watkins P C. Rapid and efficient cloning of Alu-PCR products using uracil DNA glycosylase[J].PCR Methods Appl, 1991,1(2):121-123.
[10]Hansen N B, Lübeck M, Lübeck P S. Advancing USER cloning into simpleUSER and nicking cloning[J].J Microbiol Methods, 2014,96:42-49.
[11]Villiers B, Hollfelder F. USER friendly DNA recombination(USERec): gene library construction requiring minimal sequence homology[J].Methods Mol Biol, 2014, 1179: 213-224.
[12]Li M Z, Elledge S J. Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC[J].Nat Methods, 2007,4(3): 251-256.
[13]Li M Z, Elledge S J. SLIC: A Method for Sequence- and Ligation-Independent Cloning[J].Methods Mol Biol, 2012,852:51-59.
[14]Blanusa M, Schenk A, Sadeqhi H,etal. Phosphorothioate-based ligase-independent gene cloning(PLICing): An enzyme-free and sequence-independent cloning method[J].Anal Biochem, 2010,406(2):141-146.
[15]Gan R, Wu X, He W,etal. DNA phosphorothioate modifications influence the global transcriptional response and protect DNA from double-stranded breaks[J].Sci Rep, 2014,4:6642.
[16]Howland S W, Poh C M, Rénia L. Directional, seamless, and restriction enzyme-free construction of random-primed complementary DNA librariesusing phosphorothioate-modified primers[J].Anal Biochem, 2011,416(1):141-143.
[17]Nikiforov T T, Rendle R B, Kotewicz M L,etal. The use of phosphorothioated primers and exonuclease hydrolysis for the preparation of single-strand PCR products and their detection by solid-phase hybridization[J].PCR Methods Appl, 1994,3(5):285-291.
Research of Long Fragment Gene Cloning Using T7 Exonuclease and Phosphorothioated Primers
LAN Hong, ZHANG Yu-xiang
(Department of Biochemistry and Molecular Biology, Capital Medical University, Beijing 100069, China)
Objective To clone the long coding sequence of Notch2 using T7 exonuclease and phosphorothioated primers by the ligation-independent cloning method. Methods The long coding sequence of Notch2 (7416 bp) was artificially divided into three fragments because it was difficult for PCR amplification. The primers, which were used to amplify three Notch2 fragments and the vector backbone, were phosphorothioated respectively. The three fragments of Notch2 and the vector backbone were amplified by the primers, and the PCR products were digested by T7 exonuclease, and then four fragments with 3′ complementary single-stranded overhangs were produced. The complementary ends were annealed, and the Notch2 cloning was performed. Results The result of agarose Gel electrophoresis showed that PCR products of three Notch2 fragments and the vector backbone were consistent to the expectation in size. The recombinant of pcDNA3.0-3*Flag-Notch2 was confirmed successfully by PCR, enzyme digestion and DNA sequencing. Conclusion The long fragment gene can be cloned successfully by the ligation-independent cloning method using T7 exonuclease and phosphorothioated primers.
Notch2 gene; Long fragment; Ligation-independent cloning; T7 exonuclease; Phosphorothioated primer
100069 北京,首都医科大学生物化学与分子生物学系
R349.61
A
2095-140X(2015)08-0051-05
10.3969/j.issn.2095-140X.2015.08.013
2015-05-16 修回时间:2015-06-07)
猜你喜欢
中国慈善家(2022年3期)2022-06-14
中等数学(2022年1期)2022-06-05
现代苏州(2022年9期)2022-05-26
快乐语文(2021年34期)2022-01-18
中国(俄文)(2020年8期)2020-11-23
中等数学(2018年4期)2018-08-01
中学数学研究(广东)(2018年23期)2018-03-05
初中生世界·九年级(2017年9期)2017-10-13
系统工程与电子技术(2016年2期)2016-04-16
中国光学(2015年1期)2015-06-06
