
Deletion of a Non-Catalytic Region Increases the Enzymatic Activity of a β-Agarase from Flammeovirga sp. MY04
2015-03-15 01:43HANWenjunGUJingyanLIUHuihuiLIFuchuanWUZhihongandLIYuezhong
HAN Wenjun, GU Jingyan LIU Huihui LI Fuchuan, WU Zhihong and LI Yuezhong
1) State Key Laboratory of Microbial Technology, School of Life Science, Shandong University, Jinan 250100, P. R. China
2) National Glycoengineering Research Center, Shandong University, Jinan 250100, P. R. China
Deletion of a Non-Catalytic Region Increases the Enzymatic Activity of a β-Agarase from Flammeovirga sp. MY04
HAN Wenjun1),2), GU Jingyan1), LIU Huihui1), LI Fuchuan2), WU Zhihong1), and LI Yuezhong1),*
1) State Key Laboratory of Microbial Technology, School of Life Science, Shandong University, Jinan 250100, P. R. China
2) National Glycoengineering Research Center, Shandong University, Jinan 250100, P. R. China
© Ocean University of China, Science Press and Springer-Verlag Berlin Heidelberg 2015
A Glycoside hydrolase (GH) typically contains one catalytic module and varied non-catalytic regions (NCRs). However, effects of the NCRs to the catalytic modules remain mostly unclear except the carbohydrate-binding modules (CBMs). AgaG4 is a GH16 endo-β-agarase of the agarolytic marine bacterium Flammeovirga sp. MY04. The enzyme consists of an extra sugar-binding peptide within the catalytic module, with no predictable CBMs but function-unknown sequences in the NCR, which is a new characteristic of agarase sequences. In this study, we deleted the NCR sequence, a 140-amino acid peptide at the C-terminus and expressed the truncated gene, agaG4-T140, in Escherichia coli. After purification and refolding, the truncated agarase rAgaG4-T140 retained the same catalytic temperature and pH value as rAgaG4. Using combined fluorescent labeling, HPLC and MS/MS techniques, we identified the end-products of agarose degradation by rAgaG4-T140 as neoagarotetraose and neoagarohexaose, with a final molar ratio of 1.53:1 and a conversion ratio of approximately 70%, which were similar to those of rAgaG4. However, the truncated agarase rAgaG4-T140 markedly decreased in protein solubility by 15 times and increased in enzymatic activities by 35 times. The oligosaccharide production of rAgaG4-T140 was approximately 25 times the weight of that produced by equimolar rAgaG4. This study provides some insights into the influences of NCR on the biochemical characteristics of agarase AgaG4 and implies some new strategies to improve the properties of a GH enzyme.
agarase; enzymatic characteristic; Flammeovirga; non-catalytic region; oligosaccharide; truncation
1 Introduction
Glycoside hydrolases (GHs) can hydrolyze polysaccharides into oligosaccharides or monosaccharides. Typically, a GH enzyme contains a GH module and one or more non-catalytic regions (NCRs). The GH module is responsible for the binding of saccharide rings and the hydrolyzing of glycoside linkages, thus determining the substrate-degrading patterns (Cantarel et al., 2009). According to the conservation of GH modules, glycoside hydrolases have been classified into 133 families (the CAZy database: www.cazy.org). Different enzymes within a family share the same conserved active site residues in their GH modules, but contain specific NCR sequences. Several sequence characteristics and functions have been described in NCRs, such as carbohydrate binding modules (CBMs), thrombospondin type 3 repeats (TSP3), and bacterial immunoglobulin-like domains of group 2 (Big2 domains) (Bourne and Henrissat, 2001; Cantarel et al., 2009; Michel et al., 2009). The CBMs inthe non-catalytic regions are able to form substrate-binding grooves to help enzymes bind substrates more efficiently in some GH enzymes such as agarases (Henshaw et al., 2006), cellulases (Arai et al., 2003; Boraston et al., 2004) and xylanases (Mamo et al., 2007). However, effects of other types of NCRs on enzymatic properties remain mostly unclear.
Agarose is the polysaccharide of repeated disaccharide units of 3,6-anhydro-L-galactopyranose-α-1,3-D-galactose, linked by β-1,4 bonds, which are the major polysaccharide constituent of most red algae (Rhodophydta) (Chi et al., 2012; Fu et al., 2010; Michel et al., 2006; Rees, 1969). Agarases are those GH enzymes responsible for the degradation of agarose. According to the glycoside linkages they hydrolyze, agarases are classified into two groups. The α-agarases act on the α-1,3 linkages in agarose, thus producing agaro-oligosaccharides with 3,6-anhydro-L-galactopyranose as the reducing end (Rochas et al., 1994), while the β-agarases cleave the β1-4 linkages to produce neoagaro-oligosaccharides with D-galactose as the reducing end (Morrice et al., 1983).
We have recently cloned an endo-β-agarase gene (agaG4) from an agarolytic marine bacterium, Flammeo-virga sp. MY04 (Han et al., 2012). The product of agaG4 contains a Type I signal peptide at the N-terminus, followed by a GH16 module (Gln21-Lys363), a Ser/Gly-rich linker (Ser364-Ser370), and a putative por secretion system sorting domain (Lys436-His503) at the C-terminus. AgaG4, together with the agarases AgaYT of F. yaeyamensis strain YT (Yang et al., 2012), AgaD of Z. galactanivorans DSij (Hehemann et al., 2013), and the predicated protein MS116 of Microscilla sp. PRE1 (Zhong et al., 2001), forms a new subclass of the GH16 family, due to a particularly conserved extra peptide in their catalytic modules (Han et al., 2013). Truncation of this peptide from AgaG4 decreased the protein solubility and the optimal catalytic temperature of the truncated protein, but increased the enzymatic activity and changed the substrate degradation pattern (Han et al., 2013). Another sequence characteristic of the four subfamily members is that, only the MS116 protein contains a CBM6-like domain while the other three have no predictable CBMs within the NCRs. In this study, we investigated the effects of the special NCR fragment of AgaG4 on the properties of the catalytic module.
2 Materials and Methods
2.1 Bacterial Strains, Plasmids and Chemicals
The marine agarolytic bacterium Flammeovirga sp. MY04 (CGMCC 2777) was cultivated at 30℃ in a broth (pH 7.2) containing 0.75% KCl, 3.0% NaCl, 0.11% CaCl2, 0.72% MgSO4, 0.40% tryptone, and 0.25% yeast extract. Escherichia coli strains were cultivated at 37℃ in Luria-Bertani (LB) broth. Agar (1.5%) was added into the medium to prepare the corresponding solid media. Ampicillin was supplemented at a final concentration of 100 μg mL-1when required. Genes were expressed following a PBADpromoter in the expression vector pBAD/gIII A (Novagen, USA). Plasmids were prepared and purified using the TIANpure Mini Plasmid Kit (Tiangen Co. Ltd, Beijing, China) according to the manufacturer’s manual. The PrimeSTARTMHS DNA polymerase, T4 polynucleotide kinase, and T4 DNA ligase were purchased from Ta-KaRa Corporation (Dalian, China). All other chemicals used in this study were of commercially available analytical grade.
2.2 Construction of Expression Vectors
The plasmid pBAgaG4 encodes the full-length protein of AgaG4 (rAgaG4) (Han et al., 2013). To delete the NCR (Ser364-His503) fragment from AgaG4, the plasmid pBAgaG4 was used as the DNA template in the inverse PCR amplification. The primer pair of GH16End-r (5’-CTTAGGTTTGTAAACCCTCACCCAATCTACC-3’) and PXbaI-f
(5’-TTTCTAGAACAAAAACTCATCT CAGAAGAGG-3’) was designed to target the 3’-sequences of the GH16 module-encoding region and the sequences from the XbaI inserting site of pBAgaG4, respectively. The DNA amplification products were gel recovered, end phosphorylated, and self-circularized (pBAgaG4-T140). The recombinant plasmids were sequenced to confirm the integrity of nucleotide sequences.
2.3 Heterologous Expression and Purification of rAgaG4-T140
We expressed and purified the recombinant protein rAgaG4-T140 using a similar strategy previously described for rAgaG4 (Han et al., 2013). Briefly, E. coli TOP10 cells harboring the plasmid pBAgaG4-T140 were cultivated in LB broth to a cell density of 0.8 (A600) and induced to produce the recombinant protein by adding L-arabinose. The cells were harvested by centrifugation (6 000×g, for 15 min), washed twice using ice-cold buffer A (50 mmol L-1Tris, 150 mmol L-1NaCl, pH 8.0), resuspended in buffer A, and disrupted by sonification (60 repetitions, 5s) in an ice-water bath. After centrifugation, the precipitations were collected and washed twice in buffer A supplemented with 1.0% Triton X-100 and 2 mol L-1urea, respectively. The partially purified rAgaG4-T140 inclusion bodies were dissolved in buffer B (buffer A supplemented with 8 mol L-1urea) at 4℃. The recombinant protein was purified using a nickel-nitrilotriacetic acid (Ni-NTA) column (Novagen, USA) and then eluted using buffer B containing additional imidazole at increasing gradient concentrations (0, 10, 50, and 250 mmol L-1). Fractionated protein samples were analyzed using a 13.2% SDS-PAGE (sodium dodecyl sulfatepolyacrylamide gel electrophoresis) and stained with Coomassie Brilliant Blue R-250 (Sambrook and Russel, 2001). The purified 40-kDa protein solution was diluted and refolded by dialyzing against the universal TGE buffer (50 mmol L-1Tris, 50 mmol L-1NaCl, 0.5 mmol L-1EDTA, 5 mmol L-1DTT, 5% glycerol, pH 7.9), supplemented with 4 mol L-1urea and then 2 molL-1urea and finally without urea. To completely remove the urea, the enzyme solution of rAgaG4-T140 was dialyzed against the TGE buffer (1:50) for 2 h at 4℃ for a total of 4 times. The unsuccessfully refolded proteins were removed from the dialyzed mixture by centrifugation (15 000×g, for 30 min) (Coligan et al., 2003). Protein concentrations in the study were determined using Folin Ciocalteu’s phenol reagent (Sigma-Aldrich, USA) according to the manufacturer’s protocol, with bovine serum albumin as a standard.
2.4 Biochemical Characterization of rAgaG4-T140
The agarase activity of rAgaG4-T140 was assayed using the 3,5-dinitrosaliclic acid method as described for rAgaG4 (Han et al., 2013; Miller, 1959). The absorbance of reducing sugars was measured at 540 nm and compared with the standard curve of D-galactose. One unit of enzyme was defined as the amount of enzyme that produced 1 μmol of D-galactose per minute.
The substrate specificity of rAgaG4-T140 was determined for 12 h at different temperatures from 20℃ to 50℃, using 0.10% agar, agarose, alginate, carrageenan, and ι-carrageenan as substrates. To determine the optimal conditions for rAgaG4-T140 activity, stock solutions ofagarose (0.10%) were prepared using buffers of different pH values, including the acetate buffer (50 mmol L-1, pH 4.0-6.5), the HEPES buffer (50 mmol L-1, pH 6.0-8.0), and the Tris-HCl buffer (50 mmol L-1, pH 7.0-10). The optimal temperature of rAgaG4-T140 was assayed by monitoring the agarase activity at temperatures ranging from 0 to 80℃, at pH 7.5 for 1 h. The pH dependence of rAgaG4-T140 was tested at 50℃ for 1 h.
The effects of metal ions and chelating agents on rA-gaG4-T140 activity were assayed by determining the agarose-degrading activity in the presence of the compounds at final concentrations of 1 and 10 mmol L-1, respectively.
2.5 Analysis of the Agarose-Degrading Properties of rAgaG4-T140
The agarose (0.05) was digested by rAgaG4-T140 (5 U L-1) at 45℃ and traced over 72 h. Aliquots of the reaction product were collected for time course analysis by thin layer chromatography (TLC) using silica gel 60 plates (F254, Merck). After being loaded with the oligosaccharide samples (1 μg each), each plate was developed using the solvent system containing n-butanol/ethanol/water at a volume ratio of 2:1:1. To visualize the oligosaccharide spots, the developed plates were spayed using a staining solution (1.0% diphenylamine and aniline in acetone) and heated at 110℃ for 10 min. Standard neoagaro-oligosaccharides containing neoagarobiose (NA2), neoagarotetraose (NA4), neoagarohexaose (NA6), and neoagarooctaose (NA8) were prepared using the β-agarase AgaB from Pseudoaltermonas sp. CY24 (Ma et al., 2007). Oligosaccharide concentrations of the degradation products were determined using the dinitrosa-licylic acid reducing sugar assay (Miller, 1959).
To determine the molecular masses, oligosaccharides in the final agarose degradation products of rAgaG4-T140 were purified using the Superdex Peptide 10/300 GL column (GE Healthcare, USA), monitored by a parallax detector (Shimadzu, Kyoto, Japan). The mobile phase was a NH4HCO3solution (0.10 mol L-1) at a flow rate of 0.4 mL min-1. Fractionized samples containing oligosaccharide monomers were concentrated by repeating rotary evaporation to remove salts. Pure oligosaccharides (approximately 5 μg) were assayed using MALDI-TOF MS (AXIMA-CFR plus, Shimadzu, Japan).
To judge the saccharide units at the reducing ends, oligosaccharide mixtures (approximately 5 μg) in the final agarose degradation products of rAgaG4-T140 were labeled using superfluous 2-aminobenzamide (2-AB) and sodium cyanoborohydride reagents as described by Bigge et al. (1995). The fluorescently labeled products were analyzed using a two-stage MS spectrometry. To determine the oligosaccharide compositions, the 2-AB labeled products (0.05 μg per sample) were analyzed by an HPLC system (Shimadzu Ltd., Kyoto, Japan) using a BioSep SEC-S2000 column (300 mm×7.8 mm, Phenomenex, Torrance, CA, USA). The samples were stimulated at 330 nm and monitored at 420 nm. The mobile phase was a NH4HCO3solution (0.10 mol L-1) at a flow rate of 0.5 mL min-1. The online monitoring and analysis of the data were performed using the Shimadzu LC solution software package (version 1.25).
2.6 Degradation Properties of Oligosaccharides by rAgaG4-T140
Agarose was partially digested by the agarase rAgaG4 to produce neoagaro-oligosaccharides (e.g., NA4, NA6, NA8 and NA10). Oligosaccharide mixtures in the enzymatic products were purified using the Superdex Peptide 10/300 GL column. Each purified oligosaccharide was mixed with rAgaG4-T140 (0.10 U mL-1) at a final concentration of 0.05%, and the mixture was incubated at 45℃ for 12 h. The enzymatic reaction was stopped by heating the mixtures in boiling water for 10 min and then cooled at 4℃. After centrifugation at 15 000×g for 15 min, the supernatant of the product mixtures was collected as the final degradation products of oligosaccharides. To assay the smallest substrate and the minimum degradation products, each oligosaccharide substrate and the final degradation products were directly analyzed using the TLC method. To determine the molar ratios, each oligosaccharide substrate and its final degradation product were labeled at the reducing ends with superfluous 2-AB, and then analyzed using the fluorescent HPLC system.
To determine the oligosaccharide-degrading pattern, the purified oligosaccharides were initially labeled using superfluous 2-AB, then purified using a BioSep SECS2000 column, and finally digested with rAgaG4-T140. The final degradation products and the negative samples were analyzed using the fluorescent HPLC system.
3 Results
3.1 Gene Truncation, Heterologous Expression, and Purification of rAgaG4-T140
The NCR fragment (Ser364-His503) of AgaG4 contains a putative C-domain associated with the outer membrane and covalent modification (Han et al., 2013). This is a new molecular organization type of NCR in the GH16 agarases. We truncated the NCR-encoding sequence of the agaG4 gene from the plasmid pBAgaG4, generating a new plasmid, named pBAgaG4-T140. The GH16 module (Gln21-Lys363) was completely retained in rAgaG4-T140. Similar to the full-length rAgaG4 protein, the truncated protein (rAgaG4-T140) contained a gIII virus signal peptide at the N-terminus and a 6×His tag at the C-terminus (Fig.1). The predicted molecular mass of the engineered protein was 40 kDa, approximately 15 kDa smaller than that of rAgaG4. The predicted pI value of rAgaG4-T140 is 6.04, little different from that of rAgaG4 (6.25).

Fig.1 Schematic of rAgaG4 (A) and rAgaG4-T140 (B). The non-catalytic region was deleted from rAgaG4 to form the rA-gaG4-T140 protein that contains an extra peptide (in green) of 57-amino acid residues within the GH16 module (in blue).
SDS-PAGE analysis showed that the cells could hardly produce soluble products but form inclusion bodies of rAgaG4-T140, with the correct molecular mass of approximately 40 kDa. The cells produced more than 850 mg L-1inclusion bodies after being induced by L-arabinose at a final concentration ≥ 1.0 mmol L-1. To obtain active agarases, rAgaG4-T140 in the inclusion bodies were dissolved using urea-contained buffer, purified by Ni-NTA affinity chromatography. SDS-PAGE analysis showed that denatured rAgaG4-T140 proteins were eluted from the Ni-column by imidazole at concentrations ranging from 50 to 150 mmol L-1, with a purity > 95% and a recovery of approximately 75%. When dialyzed against the TGE buffer (pH 7.9) at initial protein concentrations below 10 μg mL-1, the purified rAgaG4-T140 proteins (Fig.2) were refolded successfully. However, the solubility of rAgaG4-T140 was only about 1/15 that of rAgaG4. Thus, the dialyzed enzymes were used for further enzymatic assays after centrifugation.
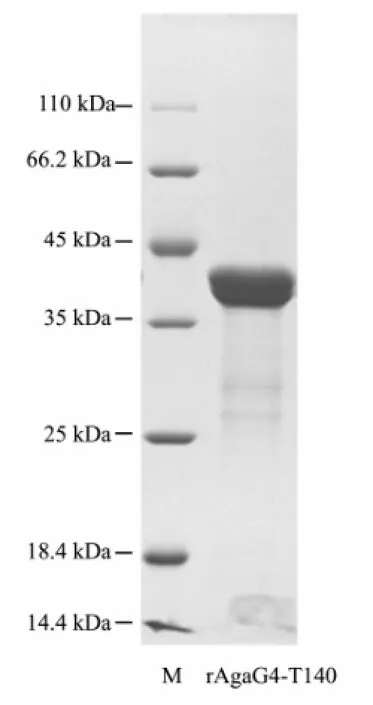
Fig.2 SDS-PAGE analysis of the purified protein of rA-gaG4-T140. M, standard protein markers. rAgaG4-T140, the protein purified from the Ni-NATA column.
3.2 Enzymatic Characteristics of rAgaG4-T140
Similar to rAgaG4, the refolded rAgaG4-T140 protein was able to hydrolyze agar and agarose but not alginate, carrageenan, or ι-carrageenan. The optimal temperature and pH value for the agarose degradation by rAgaG4-T140 were 50℃ and 7.5 respectively, the same as for rAgaG4. The protein showed more than 85% relative activities after 1 h of enzymatic reactions at various temperatures ranging from 40℃ to 50℃, but lost activity when temperatures were higher than 60℃ (Fig.3A). The agarase showed more than 80% relative activities after 1 h of enzymatic reactions in environments of different pH values ranging from 6.5 to 8.0, but showed weak activity if at pH values below 5 (Fig.3B). The agarase activity of rAgaG4-T140 was completely inhibited by Ag+, Hg2+, or Pb2+and slightly inhibited by Ni2+, Zn2+, Fe3+, EDTA, SDS, or cetyltrimethyl ammnium bromide. In contrast, the agarase activity of rAgaG4-T140 was slightly increased by the ions of Na+, Li+, Ca2+, or Mn2+. Notably, DTT (10 mmol L-1) increased the agarase activity up to 12 folds of the initial value. Surprisingly, under the optimal conditions of 50℃ and pH 7.5, the specific agarase activity of rAgaG4-T140 was approximately 5 000 U mg-1proteins, almost 35 times of the enzyme activity of rAgaG4.

Fig.3 Effects of temperatures (A) and pH values (B) on the agarase activity of rAgaG4-T140. The results are means ± S.D. of three different experiments.
3.3 Agarose Degradation Properties of rAgaG4-T140
Similar to the endo-type agarase rAgaG4 (Han et al., 2013), the agarase rAgaG4-T140 degraded agarose into oligomers progressively, and finally produced two oligosaccharides as the end products, which showed Rfvalues identical to those of NA6 and NA4, respectively (Fig.4A). After purification, the two oligosaccharide-products showed molecular masses of agarose-derived hexasaccharide (936) and tetrasaccharide (630) in the MS analysis. To determine the reducing ends of the oligosaccha-ride-products, instead of using the typical13C-NMR method, we labeled the oligosaccharide mixtures using 2-AB to produce asymmetric molecules and analyzed the products using a two-stage MS spectrometry. The MS/MS analysis showed that the saccharide sequence of the 2-AB labeled tetra-saccharide was ‘AGAG-2-AB’ rather than‘GAGA-2AB’ (Fig.5). This result indicated that the products were neoagaro-oligosaccharides containing D-galactose as the reducing ends. The truncated protein rAgaG4-T140 acted as an endo-β-agarase in agarose degradation.
To identify the oligosaccharide compositions, the final agarose degradation products were labeled by 2-AB and analyzed using a fluorescent HPLC system. The results showed that NA4 and NA6 accounted for more than 99% of the final agarose degradation products at a final molar ratio of 1.53:1 (Fig.4B). When 0.05% agarose was completely digested by rAgaG4-T140, approximately 0.5 mmol L-1reducing oligosaccharides were produced, detected using the dinitrosalicylic acid-reducing sugar assay. The yield level and the compositions of the oligosaccharide products were the same even if agarose increased to a concentration of 0.25% or rAgaG4-T140 decreased to a final concentration of 0.20 U mL-1. These results indicate that the truncated agarase rAgaG4-T140 was able to completely degrade agarose and produce equal weights of NA4 and NA6 as the final products, at a conversion ratio of approximately 70% (oligosaccharide/agarose). The neoagaro-oligosaccharides produced by rAgaG4-T140 were approximately 25 times heavier than those produced by equimolar rAgaG4 (Han et al., 2013).

Fig.4 TLC detection (A) of the agarose-degradation products by rAgaG4-T140 and fluorescent-HPLC analysis (B) of the final oligosaccharide products labeled by 2-AB. M, standard markers of neoagaro-oligosaccharides ranging from NA2 to NA8. 2-AB, 2-amino benzamide. The molar ratio of NA4 and NA6 was determined to be 1.53:1 according to the peak area.
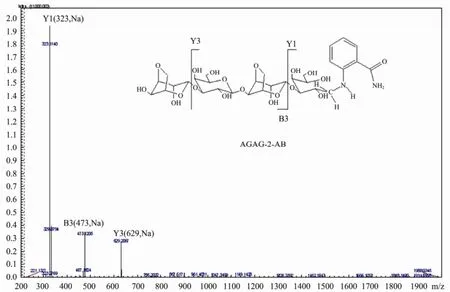
Fig.5 MS/MS analysis of the sequence of 2-AB labeled neoagarotetraose produced by rAgaG4-T140. A, 3,6-anhydro-L-galactopyranose; G, α-1,3-D- galactose.
3.4 NAOs Degradation Properties of rAgaG4-T140
To determine the smallest substrate and the minimum oligosaccharide product, the NAOs monomers including NA4, NA6, NA8, and NA10 were each digested by the agarase rAgaG4-T140, respectively. The final enzymatic products were directly assayed using silica plates, and fluorescently analyzed using a HPLC system after the 2-AB labeling. TLC analysis revealed that rAgaG4-T140 did not degrade NA6 or NA4 but digested NA8 to produce NA4 and hydrolyzed NA10 to produce NA6 and NA4 (Fig.6). Fluorescent HPLC analysis indicated that rAgaG4-T140 divided NA8 into double NA4 molecules (Fig.7A) and degraded NA10 into equimolar NA6 andNA4 (Fig.7B). Therefore, NA8 was the smallest substrate of rAgaG4-T140 and NA4 was the minimum oli-gosaccharide product.

Fig.6 TLC analyses of the oligosaccharides degradation by the agarases rAgaG4 (G4) and rAgaG4-T140 (T140). M, standard markers of neoagaro-oligosaccharides ranging from NA2 to NA8. (-), negative control group. G4, the degradation products by rAgaG4. T140, by rAgaG4-T140.
To determine the degradation patterns, the oligosaccharides NA8 and NA10 were labeled using superfluous 2-AB, purified into monomers, and then digested by rA-gaG4-T140. The fluorescent HPLC analysis indicated that rAgaG-T140 digested 2-AB-labeled NA8 to produce equimolar 2-AB-labeled NA4 and unlabeled NA4 (Fig. 7C). When rAgaG4-T140 degraded 2-AB-labeled NA10, it yielded 2-AB-labeled NA6 and unlabeled NA4 (Fig. 7D). The results showed that the minimum oligosaccharide product (NA4) of rAgaG4-T140 was cleaved from the non-reducing end of substrates.
4 Discussion
Functions of NCRs in an enzyme are mysterious. Most of the elucidated agarases, the α- or β-type, contain one or more CBM domains in their non-catalytic regions (Chi et al., 2012; Fu et al., 2012; Michel et al., 2006). The CBM domains in agarase are conserved in amino acid sequences and coevolved with the catalytic modules (Michel et al., 2009). For example, the CBM6 domains in agarases AgaA and AgaB of Zobellia galactanivorans DSij are able to bind to the non-reducing end of agarose specifically and help the agarases to hydrolyze substrates efficiently (Henshaw et al., 2006). The β-agarases AgaA of Pseudoalteromonas sp. CY24 (GenBankTMAY150179) and AgaD of Vibrio sp. PO-303 (GenBankTMAB221476), which share 98% similarity of the protein sequences, contain CBM domains belonging to the CBM13 family (Lu et al., 2009; Dong et al., 2007). Deletion of the CBM13 domain from AgaD led to an increase in the enzyme activity of the truncated protein (Dong et al., 2007). Besides the CBM domains, some other types of module, such as TSP3, Big2, and modules of unknown functions were also found in the NCRs of many agarases (Michel et al., 2009). However, the influences of non-typical NCR modules on the enzymatic properties of agarases have not yet been elucidated.
AgaG4 contains no predicable CBM domains in the NCR. While the extra peptide in the GH16 module of AgaG4 was demonstrated to affect many enzymatic properties (Han et al., 2013), the function of NCR remains unclear. In this study we showed that the deletion of the NCR fragment from rAgaG4 did not affect the characteristics of the enzyme in many aspects. For example, like the original protein rAgaG4, rAgaG4-T140 degraded agarose, producing NA4 and NA6 as end products at a final molar ratio of approximately 1.5:1, and the smallest substrate of rAgaG4-T140 was NA8 and the minimum product was NA4, departed from the non-reducing end of substrates. The optimal temperature and pH value for agarolytic activity of rAgaG4-T140 were also the same (50℃ and pH 7.5) as rAgaG4. The effects ofmetal ions and chelating agents were similar on rAgaG4-T140 and rAgaG4. These results indicate that the GH16 module of AgaG4 is the key domain while the NCR fragment is not required for determining enzymatic characteristics and substrate-degrading properties. However, the initial protein concentration for successful refolding and the enzyme activity of refolded rAgaG4-T140 were 1/15 and 35 times those of rAgaG4, respectively. The neoagaro-oligosaccharides produced by rAgaG4-T140 are approximately 25 times the weight of that produced by equimolar rAgaG4. The results show that deletion of the NCR from the agarase AgaG4 has decreased protein solubility but increased enzyme activity and the oligosaccharide productivity.
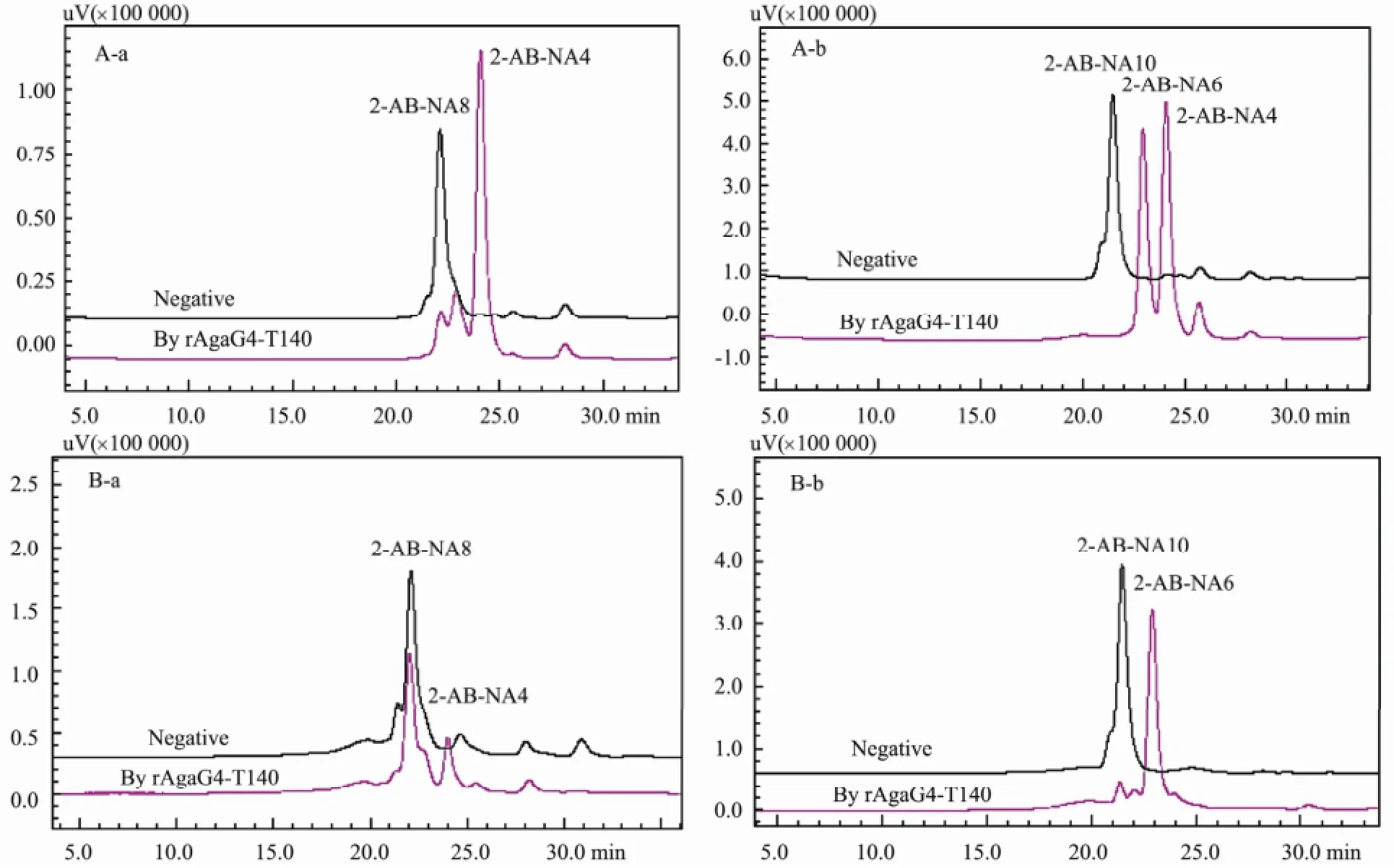
Fig.7 Fluorescent HPLC analysis of the 2-AB labeled oligosaccharide- degradation products by rAgaG4-T140. Natural oligosaccharides NA8 (A-a) and NA10 (A-b) as substrates; 2-AB labeled oligosaccharides NA8 (B-a) and NA10 (B-b) as substrates.
Agarases have been extensively investigated because there are many functions of agarose-derived oligosaccharides, such as anti-oxidation (Wang et al., 2004; Wu et al., 2005), anti-inflammation (Higashimura et al., 2013), and prebiotic effects (Hu et al., 2006). The monosaccharide 3,6-anhydro-L-galactopyranose also showed obvious skin whitening and anti-inflammatory activities (in vitro) with no significant cytotoxicity (Yun et al., 2013). However, mainly due to their low efficiency in agarose degradation, natural agarases are normally unsatisfactory in industrial preparation of agarose oligosaccharides. To improve the enzyme properties, two wild-type agarases were modified by random mutations via error-prone PCR or DNA shuffling (Jang et al., 2010; Shi et al., 2008). Although these two studies have succeeded in increasing the thermostabilities of agarases, the enzyme activities and other properties have been poorly improved (Lee et al., 2011). The studies present in this paper not only provide insights into the influences of the NCR fragments on the biochemical characteristics of agarase AgaG4, but also imply some new strategies to improve the properties of an enzyme.
Acknowledgements
This work was financially supported by the Open Research Fund Program of Shandong Provincial Key Laboratory of Glycoscience & Glycotechnology (Ocean University of China) KLGG (OUC) 201301, the National Natural Science Foundation of China Grants 31300664 and 31130004, and the State Key Laboratory of Microbial Technology Grant (Shandong University) M2013-11.
Arai, T., Araki, R., Tanaka, A., Karita, S., Kimura, T., Sakka, K., and Ohmiya, K., 2003. Characterization of a cellulase containing a family 30 carbohydrate-binding module (CBM) derived from Clostridium thermocellum CeIJ: Importance of the CBM to cellulose hydrolysis. Journal of Bacteriology, 185: 504-512.
Bigge, J. C., Patel, T. P., Bruce, J. A., Goulding, P. N., Charles, S. M., and Parekh, R. B., 1995. Nonselective and efficient fluorescent labeling of glycans using 2-amino benzamide and anthranilic acid. Analytical Biochemistry, 230: 229-238.
Boraston, A. B., Bolam, D. N., Gilbert, H. J., and Davies, G. J., 2004. Carbohydrate-binding modules: fine-tuning polysac-charide recognition. Biochemical Journal, 382: 769-781.
Bourne, Y., and Henrissat, B., 2001. Glycoside hydrolases and glycosyltransferases: families and functional modules. Current Opinion of Structural Biology, 11: 593-600.
Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B., 2009. The carbohydrate- active enzymes database (CAZy): An expert resources for glycogenomics. Nucleic Acids Research, 37: D233-D238.
Chi, W. J., Chang, Y. K., and Hong, S. K., 2012. Agar degradation by microorganisms and agar-degrading enzymes. Applied Microbiology and Biotechnology, 94: 917-930.
Coligan, J. E., Dunn, B. M., Speicher, D. W., and Wingfield, P. T. 2003. Short Protocols in Protein Science. Willey, New York, 1-78.
Dong, J., Tamaru, Y., and Araki, T., 2007. Molecular cloning, expression, and characterization of a beta-agarase gene, agaD, from a marine bacterium, Vibrio sp. strain PO-303. Bioscience Biotechnology and Biochemistry, 71: 38-46.
Fu, X. T., and Kim, S. M., 2010. Agarase: Review of major sources, categories, purification method, enzyme characteristics and applications. Marine Drugs, 8: 200-218.
Han, W. J., Gu, J. Y., Liu, H. H., Li, F. C., Wu, Z. H., and Li, Y. Z., 2013. An extra peptide within the catalytic module of a β-agarase affects the agarose degradation pattern. The Journal of Biological Chemistry, 288: 9519-9531.
Han, W. J., Gu, J. Y., Yan, Q. J., Li, J. G., Wu, Z. H., Gu, Q. Q., and Li, Y. Z., 2012. A polysaccharide-degrading marine bacterium Flammeovirga sp. MY04 and its extracellular agarase system. Journal of Ocean University of China, 11: 375-382.
Hehemann, J. H., Correc, G., Thomas, F., Bernard, T., Barbeyron, T., Jam, M., Helbert, W., Michel, G., and Czjzek, M., 2012. Biochemical and structural characterization of the complex agarolytic enzyme system form the marine bacterium Zobellia galactanivorans. The Journal of Biological Chemistry, 287: 30571-30584.
Henshaw, J., Horne-Bitschy, A., Bueren, A. L., Money, V. A., Bolam, D. N., Czjzek, M., Ekborg, N. A., Weiner, R. M., Hutcheson, S. W., Davies, G. J., Boraston, A. B., and Gilbert, H., 2006. Family 6 carbohydrate binding modules in β-agarases display exquisite selectivity for the non-reducing termini of agarose chains. The Journal of Biological Chemistry, 281: 17099-17107.
Higashimura, Y., Naito, Y., Takagi, T., Mizushima, K., Hirai, Y., Akihito, H., Ohnogi, H., Yamaji, R., Inui, H., Nakano, Y., and Yoshikawa, T., 2013. Oligosaccharides from agar inhibit murine intestinal inflammation through the induction of heme oxygenase-1 expression. Journal of Gastroenterology, 48: 897-909.
Hu, B., Gong, Q. H., Wang, Y., Ma, Y. M., Li, J. B., and Yu, W. G., 2006. Prebiotic effects of neoagaro-oligosaccharides prepared by enzymatic hydrolysis of agarose. Anaerobe, 12: 260-266.
Jang, M. K., Lee, S. W., Lee, D. G., Kim, N. Y., Yu, K. H., Jang, H. J., Kim, S., Kim, A., and Lee, S. H., 2010. Enhancement of the thermostability of a recombinant β-agarase, AgaB, from Zobellia galactanivorans by random mutagenesis. Biotechnology Letters, 32: 943-949.
Lee, S., Lee, D. G., Jang, M. K., Jeon, M. J., Jang, H. J., and Lee, S. H., 2011. Improvement in the catalytic activity of β-agarase AgaA from Zobellia galactanivorans by site-directed mutagenesis. Journal of Microbiology and Biotechnology, 21: 1116-1122.
Lu, X. Z., Chu, Y., Wu, Q. Q., Gu, Y. C., Han, F., and Yu, W. G., 2009. Cloning, expression and characterization of a new agarase-encoding gene from marine Pseudoalteromonas sp. Biotechnology Letters, 31: 1665-1570.
Ma, C. P., Lu, X. Z., Shi, C., Li, J. B., Gu, Y. C., Chu, Y., Han, F., Gong, Q. H., and Yu, W. G., 2007. Molecular cloning and characterization of a novel β-agarase, AgaB, from marine Pseudoalteromonas sp. CY24. The Journal of Biological Chemistry, 282: 3747-3754.
Mamo, G., Hatti-Kaul, R., and Mattiasson, B., 2007. Fusion of carbohydrate binding modules from Thermotoga neaplitana with a family 10 xylanase from Bacillus halodurans S7. Extremophiles, 11: 169-177.
Michel, G., Barbeyron, T., Kloareg, B., and Czjzek, M., 2009. The family 6 carbohydrate-binding modules have coevolved with their appended catalytic modules toward similar substrate specificity. Glycobiology, 19: 615-623.
Michel, G., Nyval-Collen, P., Barbeyron, T., Czjzek, M., Helbert, W., 2006. Bioconversion of red seaweed galactans: A foucus on bacteria agarases and carrageenases. Applied Mcirobiology and Biotechnology, 71: 23-33.
Miller, G. L., 1959. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Analytical Chemistry, 21: 426-428.
Morrice, L. M., Mclean, M. W., Long, W. F., and Williamson, F. B., 1983. β-agarase I and II from Pseudoalteromonas atlantica: Substrate and specificites. European Journal Biochemistry, 137: 149-154.
Rees, D. A., 1969. Structure, conformation, and mechanism in
the formation of polysaccharide gels and networks. Advances
in Carbohydrate Chemistry and Biochemistry, 24: 267-332. Rochas, C., Potin, P., and Kloareg, B., 1994. NMR spectroscopic investigation of agarose oligomers produced by an α-agarase. Carbohydrate Research, 253: 69-77.
Sambrook, J., and Russel, D. W., 2001. Molecular Cloning: A Laboratory Manual. 3rd edition. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, A8.40-A8.47.
Shi, C., Lu, X. Z., Ma, C. P., Fu, X. T., and Yu, W. G., 2008. Enhancing the thermostability of a novel β-agarase AgaB through directed evolution. Applied Biochemistry and Biotechnology, 151: 51-59.
Wang, J. X., Jiang, X. L., Mou, H. J., and Guan, H. S., 2004. Anti-oxidation of agara oligosaccharides produced by agarases from a marine bacterium. Journal of Applied Phycology, 16: 333-340.
Wu, S. C., Wen, T. N., and Pan, C. L., 2005. Algal-oligosaccharide-lysates prepared by two bacterial agarases stepwise hydrolyzed and their anti-oxidative properties. Fisheries Science, 71: 1149-1159.
Yang, J. L., Chen, L. C., Shih, Y. Y., Hsieh, C. Y., Chen, C. Y., Chen, W. M., and Chen, C. C., 2012. Cloning and characterization of β-agarase AgaYT from Flammeovirga yaeyamensis strain YT. Journal of Bioscience and Bioengineering, 112: 225-232.
Yun, E. J., Lee, S., Kim, J. H., Kim, B. B., Kim, H. T., Lee, S. H., Pelton, J. G., Kang, N. J., Choi, I. G., and Kim, K. H., 2013. Enzymatic production of 3, 6-anhydrol-L-galactose from agarose and its purification and in vitro skin whitening and anti-inflammatory activities. Applied Microbiology and Biotehnology, 97: 2961-2970.
Zhong, Z. P., Toukdarian, A., Helinski, D., Knauf, V., Sykes, S., Wilkinson, J. E., O’Bryne, C., Shea, T., Deloughery, C., and Caspi, R., 2001. Sequence analysis of a 101-kilobase plasmid required for agar degradation by a Microscilla isolate. Applied and Environmental Microbiology, 67: 5771-5779.
(Edited by Ji Dechun)
(Received November 10, 2014; revised April 19, 2015; accepted April 25, 2015)
J. Ocean Univ. China (Oceanic and Coastal Sea Research)
DOI 10.1007/s11802-015-2800-0
ISSN 1672-5182, 2015 14 (5): 841-848
http://www.ouc.edu.cn/xbywb/
E-mail:xbywb@ouc.edu.cn
* Corresponding author. E-mail: lilab@sdu.edu.cn
 Journal of Ocean University of China2015年5期
Journal of Ocean University of China2015年5期
- Journal of Ocean University of China的其它文章
- Seasonal Dynamics of Turbidity Maximum in the Muthupet Estuary, India
- Decadal Variability of Global Ocean Significant Wave Height
- An Observational and Modeling Study of Extratropical Transition of Hurricane Sandy in 2012
- The Nonlinear Bifurcation and Chaos of Coupled Heave and Pitch Motions of a Truss Spar Platform
- Beach Morphology and Coastline Evolution in the Southern Bohai Strait
- Comparison of Meiofaunal Abundance in Two Mangrove Wetlands in Tong’an Bay, Xiamen, China