
残余气体质谱仪的进样输运及其对定量分析的影响
2015-03-15 05:33刘晓洋张家良
物理实验 2015年4期
吴 聪,刘晓洋,张家良
(大连理工大学 三束材料改性教育部重点实验室,辽宁 大连 116024)

残余气体质谱仪的进样输运及其对定量分析的影响
吴聪,刘晓洋,张家良
(大连理工大学 三束材料改性教育部重点实验室,辽宁 大连 116024)
摘要:使用QMS-200残余气体质谱分析仪,分别以空气、空气/氩气混合气体为样品,研究了质谱仪的进样输运效应. 利用空气/氩气混合气体作为标准样品,进行了质谱峰强对浓度的标定,获得了分析仪对残余空气定量分析的标定关系. 研究发现:由于输运过程,注入样品由于不同成分的输运速率、排空速率不同,样品不同成分在质谱仪内的量与其原始物质的量有所不同.
关键词:残余气体分析仪;输运效应;质谱峰强
1引言
质谱技术最早用来检测和分离同位素,经过长期不断完善[1-7],现已成为一种独具优势的分析技术. 由于其具有高精度及高灵敏度的优点,作为检测和分析的主要手段已被广泛应用于多种领域[8-14]. 由于质谱仪结构复杂,操作繁琐,应用领域的操作人员在使用过程中难以精通其特点,了解其局限性,对测量中出现的一些意外结果很难快速找到原因. 因此,有必要针对不同种类质谱仪的运行特性开展针对性研究,进而积累准确分析的经验和方法,这些经验和方法对于充分发挥质谱仪的应用、分析、鉴别功能很有必要.
在低温等离子体化学反应过程中会生成多种中间和最终产物,质谱仪是识别和分析这些复杂物质成分的有效工具. 分析新生物种及其反应机理,不仅需要定性识别,更需要定量分析,因此探索利用质谱仪实现等离子体化学反应体系成分的定量诊断技术具有广阔的应用前景. 质谱分析仪配置有复杂的腔体结构、超高真空机组以及样品注入和输运通道,因此气体样品需经历缓慢的输运过程,才能在分析仪腔体内稳定分布. 从气体样品注入到稳定分布的历程中,不同时刻的质谱分布不断演变,此即进样输运过程. 在输运过程的不同阶段,掌握质谱峰相对强度的演变规律是实现气体成分定量分析的关键. 本文以四极质谱残余气体分析仪[15]为研究对象,研究其输运效应以及实现气体成分定量分析的可行性.
2实验设计与流程
图1是实验系统示意图,以QMS-200残余气体质谱分析仪为核心,配以密闭样品制备室、进样器和排空泵. 改变样品真空室的抽气速率和进气流量,可以使样品气压稳定在104~105Pa之间,所以无论样品室气压如何,进样气压都远高于质谱分析区的工作气压(10-2Pa以下). 进样气压在104~105Pa之间的改变对质谱仪内的输运行为的影响不大,因此,本文实验过程中维持样品室气压5.0×104Pa不变. 为了减小样品室内残余空气的影响,注入样品气体之前,先将样品室抽至真空度为0.1 Pa. 样品室气压同样用真空计指示,调节质量流量控制器可以改变样品气成分的比例.

1.计算机 2.QMS-200质谱仪 3.密闭样品室 4.MT50质量流量控制计 5.样品气瓶 6.ZDY-54真空计 7.放气阀 8.真空泵图1 实验系统示意图
残余气体分析仪的标称指标如下:质量数范围0~200,分辨率0.1,全谱扫描时间为2 s. 对于氮分子,其检测下限为10-7Pa. 实验数据采集过程中,由于质谱扫描和进样阀开启等操作需要时间,为避免干扰,研究输运过程时,2次质谱相邻采集操作的时间间隔应大于上述时间,本文设定为10 min.
3研究结果及讨论
3.1 气体样品的质谱全图


图2 空气/氩气(1∶1混合)混合样品质谱图(样品室气压5×104 Pa,进样后立刻测量)
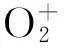
3.2 不同样品的进样输运过程
分别以大气(N278%+O221%+稀有气体1%的混合气,N2∶O2为3.7∶1),合成空气(N279%+O221%的混合气,物质的量之比为3.8∶1)和3种空气/氩气混合气(物质的量比分别为1∶1,1∶1.5,1∶2)为样品,进行输运过程实验.
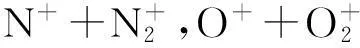

图3 合成空气样品的氮/氧质谱峰强分压 与输运时间的关系

图4 合成空气样品的氮/氧质谱峰强分压占比 与输运时间的关系
对比图3和4中可以看到,随着输运的深入,质谱中氮气和氧气峰的各自总分压持续增加,而且几乎是线性增加,氮气总分压从10 min时的9.84×10-4Pa增加到50 min时的1.86×10-3Pa,相应地,氧气总分压从1.86×10-4Pa增加到3.59×10-4Pa,均几乎增加1倍. 但二者分压占比几乎不变,为64.1%(N2)和9.5%(O2),分压比为约 6.71(见表1).
图5和6是空气/氩气1∶1混合样品的质谱峰强分压和分压占比与输运时间的关系.

图5 空气/氩气1∶1混合样品的氮/氧质谱峰强 分压与输运时间的关系
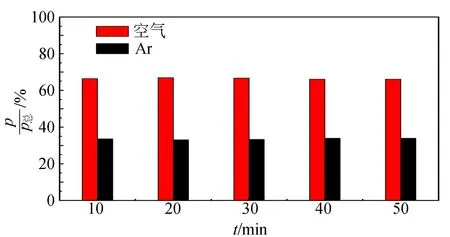
图6 空气/氩气1∶1混合样品的质谱峰强分压占比 与输运时间的关系
图5和6展现了与图3和4同样的规律性. 图5表明:对于混合样品,氮气和氧气的峰强分压随输运时间仍然保持线性增大的规律,N2从3.72×10-4Pa增加到5.32×10-4Pa,O2从6.92×10-5Pa增加到1.06×10-4Pa,二者的分压占比仍几乎不变,分别为58.1%(N2)和11.5%(O2). 由此可以计算二者分压比约为5.17(表1). 虽然氮氧的峰强分压之比不随输运时间发生变化,但是空气氩气混合样品的氮氧的峰强分压比(约5.17)略小于空气样品中的二者之比(约6.71),这表明样品的成分组成影响质谱测量. 图6显示空气与氩气的质谱峰强分压占比,也几乎不随输运时间发生变化,分别为69.6%(空气)和30.0%(氩气),二者分压占比之比约为2.28(见表1).
根据上述分析,无论在输运过程的哪一阶段进行质谱测量分析,都得到相似的结果,可以对样品气体的成分进行可信的定性分析,也可以测量出各成分的相对含量,但是不能测量绝对含量,因为不同输运阶段测量得到的成分质谱峰分压是不断变化的.
无论是合成空气样品还是混合气样品,测量得到的成分峰强分压比值并不与样品固有的物质的量比相同. 以图5为例,计算氮/氧峰强分压值比,结果见表1,可以发现氮/氧峰强分压比值(约 6.71)比合成空气中二者的物质的量比(约 3.79)大. 空气/氩气混合样品的结果也是如此,空气/氩气的峰强分压比(约2.28)比样品中二者的物质的量比(约1)大. 样品成分的质谱强度比与实际物质的量比不一致,意味着质谱峰强分压比并不能反映样品成分物质的量之比.

表1 不同样品的成分质谱峰强比值表
质谱分析仪的质谱测量过程,也是样品成分一直处于趋向稳态的动态过程. 样品气体被注入分析仪后,经历数个不同的抽空阶段,最后只有少量样品分子能够进入质谱仪的电离检测区,之后被分子泵排出. 输运过程的曲折导致样品气体最终进入电离区的概率很小,所以电离区的样品分子分压需逐渐积累升高,直至达到动态平衡. 样品分子进入电离区的速率与分子泵排出尾气的速率差别决定电离区样品分压的增长行为. 分子泵[16-17]是通过高速旋转涡轮叶片碰撞气体分子,将动量传递给气体分子,使其定向流动而实现抽气. 不同分子与涡轮叶片之间的作用不同,导致分子泵抽空不同分子的速率不同. 对比氧与氮,分子泵抽空氧分子略快,电离区内氮分子积累速率比氧分子快,因此电离区内滞留氮气的分压占比偏大. 除此之外,不同分子的电子碰撞电离截面的差别也是其质谱峰强分压占比的决定因素. 本文所用质谱仪的电离电子平均能量为70 eV,氮分子和氧分子相应的电子碰撞电离截面分别为0.017 9 nm2和0.014 9 nm2,比值约为1.20∶1. 由此可以估算得到氮分子和氧分子在电离室的积累速率之比为1.48∶1.
3.3 质谱峰强的定量标定
根据3.2的结果,残余气体分析仪得到的样品质谱强度分压占比并不能直接反映成分物质的量比. 图6展示了空气氩气(1∶1)混合气体样品的成分含量质谱分析结果,氩气与空气的峰强分压之比远小于1. 虽然按照3.2节的解释,不同气体与电子的碰撞电离截面不同[18-21]可以导致质谱峰强占比与样品物质的量不一致,但是氩原子电子碰撞电离截面(0.022 nm2)比空气主要成分(氮气和氧气0.017 9 nm2和0.014 8 nm2)分子的电子碰撞电离截面大,而图6中显示氩气的峰强分压占比远小于空气的. 因此,对于氩气空气混合样品来说,电离截面的差异不是导致氩气峰强占比偏小的唯一原因. 由此可见,对于不同的样品而言,质谱峰强占比与样品中成分物质的量的差异的原因是多样的.
为了能够定量测量气体样品成分的物质的量,需要采用标准样品对质谱峰强度进行标定. 标定程序如下:在真空样品室中,分别按照空气和氩气1∶1,1∶1.25,1∶1.5,1∶2比例混合,制备不同标准样品,并对其质谱进行测量. 前2组测量结果如图6~7所示,可发现空气与氩气的峰强分压比值仍然偏大. 3组标准样品的结果均列于表1中. 对实验测得的质谱峰强分压占比与样品实际物质的量比进行线性标定拟合,进而得到标定曲线图8,可看到,质谱峰强占比与实际物质的量比几乎成正比例关系. 标定曲线没有严格经过坐标零点,其原因在于样品室内的残余空气和质谱室内的残余氮气所致. 考虑到与零点的偏离小于10%,可以认为:通过标定程序可以得到气体样品的质谱峰强之比与样品中物质的量比的正比标定关系曲线.
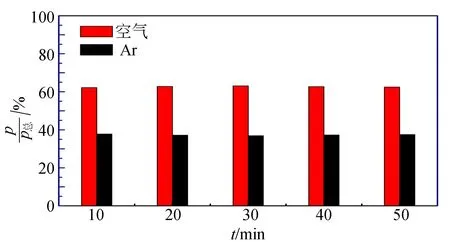
图7 空气/氩气1∶1.5样品的质谱峰强分压占比 与输运时间的关系

图8 空气/氩气质谱峰强之比与样品物质的量之比 之间的标定关系
3.4 其他较复杂结构的气体成分质谱峰强占比的标定
残余气体的检测,不仅包含Ar,N2,O2等结构简单的分子,更多的还会涉及到如H2O和CO2等结构较为复杂的分子. 图9~10是空气样品中H2O和CO22种成分的峰强分压和占比随输运时间的演变. 由图9~10可知H2O和CO2的峰强分压随输运时间的变化不同于氮氧的变化趋势,峰强分压随输运时间逐渐减小,而且不是线性的,而它们的峰强占比虽然变化幅度比峰强本身少了许多,但是仍然可以看出它们的变化,可见残余气体中微量成分的质谱峰强分压随输运过程的变化不服从主要成分N2和O2的规律. 综上可以得出结论:残余气体质谱分析仪可对残余气体主要成分定性识别,借助于正比例标定关系可以实现定量分析. 对于微量成分的定量分析由于输运现象的影响而无法直接进行.
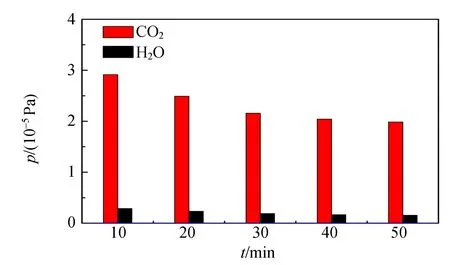
图9 空气样品中CO2和H2O质谱峰强分压 随输运时间的变化

图10 空气样品中CO2和H2O质谱峰强分压 占比随输运时间的变化
4结论
利用真空腔室作为标准样品制备室,对常规空气、合成空气、空气/氩气混合样品进行了残余气体质谱检测仪的输运过程分析. 结果表明:对于残余气体中的主要成分,经过标定,该质谱检测仪能测量样品成分的相对含量,不受输运过程的影响. 残余气体质谱仪的正比例标定过程是可行的,经标定,该分析仪可以进行样品成分物质的量的测定. 对于残余气体中的微量成分,质谱峰强度不能用于指示成分的含量,因为输运过程干扰了峰强分压与成分物质的量之间的关联性.
参考文献:
[1]Zhu L, Gamez G, Chen H W, et al. Rapid detection of melamine in untreated milk and wheat gluten by ultrasound-assisted extractive electrospray ionization mass spectrometry (EESI-MS) [J]. Chem. Commun., 2009(5):559-561.
[2]Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons [J]. Analytical Chemistry, 1988,60(20):2299-2301.
[3]Amster I J, Baldwin M A, Cheng M T, et al. Tandem mass spectrometry of higher molecular weight compounds [J]. Journal of the American Chemical Society, 1983,105(6):1654-1655.
[4]Munson M S B, Field F H. Chemical lonization mass spectrometry. I. general introduction [J]. J.Am. Chem. Soc., 1966,88(12):2621-2630.
[5] Yoshida T, Tanaka K. Sample preparation method for laser ionization mass spectrometer [P]. Japan: JP62043562-A, 1987.
[6] Yoshida T, Tanaka K. By mixing sample solution with glycerine and metal particles and applying to sample holder [P]. Japan: JP92050982-B2, 1992.
[7] Takats Z, Gologan B, Wiseman J M, et al. Desorbing and ionizing analyte in sample material in mass spectrometric analysis, comprises directing desorption electrospray ionization (DESI)-active spray droplets on sample material to desorb analyte [P].WO2005094389-A2; US2005230635-A1; EP1741120-A2; US7335897-B2; CN101073137-A; WO2005094389-A3; CA2559847-C, 2005,2007,2008,2014.
[8]Ojanperä I, Kolmonen M, Pelander A. Current use of high-resolution mass spectrometry in drug screening relevant to clinical and forensic toxicology and doping control [J]. Anal. Bioanal. Chem., 2012,403(5):1203-1220.
[9]Lee S H, Miyamoto K, Goto T, et al. Non-invasive proteomic analysis of human skin keratins: screening of methionine oxidation in keratins by mass spectrometry [J]. Journal of Proteomics, 2011,75(2):435-449.
[10]Law K P. Surface-assisted laser desorption/ionization mass spectrometry on nanostructured silicon substrates prepared by iodine-assisted etching [J]. International Journal of Mass Spectrometry, 2010,290(1):47-59.
[11]Jean-Baptisté P, Fourre E, Dapoigny A, et al. He mass spectrometry for very low-level measurement of organic tritium in environmental samples [J]. Journal of Environmental Radioactivity, 2010,101(2):185-190.
[12]Mezcua M, Ferrer C, García-Reyes J F, et al. Analyses of selected non-authorized insecticides in peppers by gas chromatography/mass spectrometry and gas chromatography/tandem mass spectrometry [J]. Food Chemistry, 2009,112(1):221-225.
[13]Ishimaru M, Yamada M, Nakagawa I, et al. Analysis of volatile metabolites from cultured bacteria by gas chromatography/atmospheric pressure chemical ionization-mass spectrometry [J]. Journal of Breath Research, 2008,2(3):037021.
[14]Huaikun L, Yaoling N. Multi-collector ICP-MS Analysis of Pb Isotope Ratios in Rocks: Data, Procedure and Caution, Acta Geologica Sinica-English Edition, 2003,77(1):44-58.
[15]Batey J H. The physics and technology of quadrupole mass spectrometers [J]. Vacuum, 2014,101:410-415.
[16]Malyshev O B. Gas dynamics modelling for particle accelerators [J]. Vacuum, 2012,86(11):1669-1681.
[17]Iqbal M, Wasy A, Batani D, et al. Design modification in rotor blade of turbo molecular pump [J]. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment,2012,678: 88-90.
[18]Yukikazu I. Cross sections for electron collisions with nitrogen molecules [J]. J. Phys. Chem. Ref. Data, 2006,35(1):31-53.
[19]Yukikazu I. Cross sections for electron collisions with oxygen molecules [J]. J. Phys. Chem. Ref. Data, 2009,38(1):1-20.
[20]Cooper J L, Pressley G A, Stafford F E. Electron impact ionization cross sections for atoms [J]. Journal of Chemical Physics, 1966,44(10):3946-3949.
[21]Hudson J E, Vallance C, Harland P W. Absolute electron impact ionization cross-sections for CO, CO2, OCS and CS2[J]. Journal of Physics B: Atomic, Molecular and Optical Physics,2004,37(2):445-455.
[责任编辑:任德香]
Sample relaxing in mass spectrometer of residual gas analysis and its effects on quantitative measurement
WU Cong, LIU Xiao-yang, ZHANG Jia-liang
(Key Laboratory for Materials Modification, Ministry of Education,
Dalian University of Technology, Dalian 116024, China)
Abstract:A QMS-200 mass spectrometry (MS) residual gas analyzer was chosen to investigate the sample relaxing process after injection. Synthetic air and air/Ar mixtures with certain ratios served as standard samples for the investigation. It was shown that MS intensities of the components did not represent the mole concentrations, because the relaxation speeds of the components were different from each other due to their different transportation and evacuation rates. For quantitative composition analysis using the analyzer, routine calibration was needed. By preparing air/Ar mixtures with different mole ratios as standard samples, the relative MS intensities of the components were found proportional to the mole ratios and therefore could be scaled for quantitative analysis.
Key words:residual gas analyzer; relaxation process; mass spectrometric intensity
中图分类号:O657.63
文献标识码:A
文章编号:1005-4642(2015)04-0012-06
作者简介:吴聪(1987-),男,河北保定人,大连理工大学物理与光电工程学院硕士研究生,研究方向为气体放电等离子体诊断.通讯作者:张家良(1967-),男,山东济南人,大连理工大学三束材料改性教育部重点实验室教授,博士,研究方向为等离子体物理.
基金项目:国家自然科学基金资助(No.11375041,10675028);中国科学院化学激光重点实验室开放基金资助(No.20131008)
收稿日期:2014-10-17;修改日期:2015-01-28
