
仙人菇口服液的质量标准研究
2015-03-09 11:51:24乐佳美熊筱娟陆文铨朴淑娟上海长征医院药学部上海00003宜春学院化学与生物工程学院江西宜春336000
中国药房 2015年27期
乐佳美,熊筱娟,陆文铨,朴淑娟,张 凤#(.上海长征医院药学部,上海 00003;.宜春学院化学与生物工程学院,江西宜春 336000)
仙人菇口服液是第二军医大学附属长征医院自制制剂,由淫羊藿、黄芪、枸杞、炒白术、人参等8味中药组成,具有益气填精、扶正固本的功效。该药主要用于慢性消耗性疾病、疲劳综合征等症,对恶性肿瘤放、化疗造成的贫血及免疫功能低下亦具有辅助的治疗作用。仙人菇口服液在本院主要应用于中晚期恶性肿瘤支持治疗,疗效确切,临床应用已近20年[1-2]。原质量标准仅有常规检查项目和对处方中淫羊藿、黄芪、枸杞的定性鉴别,尚未见有关其质量控制的文献报道。淫羊藿是仙人菇口服液的主要药材,淫羊藿苷是其有效成分之一。淫羊藿苷是一种重要的生物活性成分,能增加心脑血管血流量、促进造血功能、免疫功能及骨代谢,具有补肾壮阳、抗衰老、抗肿瘤等功效。通过攻补兼施、益气活血、扶正固本的方法,可提高中晚期恶性肿瘤患者的机体免疫机能,从而纠正患者的气血两虚的证候,稳定病情,提高生存质量。因此,为了提高产品质量的专属性、有效控制内在质量、保证临床疗效,笔者采用反相高效液相色谱(RP-HPLC)法测定淫羊藿中的淫羊藿苷的含量,为建立仙人菇口服液的质量控制方法提供参考。同时,在保留原标准的基础上增加了对处方中人参、白术的薄层色谱(TLC)鉴别,以提高军队医疗机构制剂的质量标准。
1 材料
1260 型HPLC仪,包括G1311A四元泵、G1322A真空脱气机、G1329A自动进样器、G1316A柱温箱、G4212B 二极管阵列检测器,ChemStation 软件数据处理系统(美国Agilent 公司);AG 22331 型Humberg 超高速离心机(德国Eppendorf 公司);CPA225D 型十万分之一电子分析天平(德国Sartorius 公司);WL-901 Vortex 型旋涡振荡混和器(海门市其林贝尔仪器制造有限公司);SK7200H 型高频超声波清洗器(上海科导超声仪器有限公司,功率:350 W,频率:53 kHz)。
仙人菇口服液(上海长征医院制剂中心自制,批号:130806、130807、130808,规格:10 ml/支);淫羊藿苷对照品(批号:110737~200415,纯度:98%)、黄芪甲苷对照品(批号:110781~201314,纯度:98%)、人参皂苷Rg1对照品(批号:110703~200424,纯度:98%)均由中国食品药品检定研究院提供;甲醇、乙腈、醋酸和磷酸为色谱纯,其他供薄层鉴别用试剂,包括正丁醇、甲醇、乙酸乙酯、丁酮、甲酸、氢氧化钠、氯仿、三氯甲烷、石油醚、硫酸、乙醇均为分析纯(国药集团化学试剂有限公司),水为超纯水。
2 方法与结果
2.1 TLC法定性鉴别
2.1.1 淫羊藿 取3 批样品溶液各50 ml,分别用水饱和正丁醇提取3次,每次30 ml,合并正丁醇提取液,减压浓缩至干,残渣加5 ml 甲醇溶解,作为供试品溶液。取淫羊藿苷对照品适量,加甲醇制成每ml 含1 mg 淫羊藿苷的溶液,作为对照品溶液。另取淫羊藿对照药材10 g,加甲醇50 ml,超声处理30 min,滤过,滤液用水饱和正丁醇提取2 次,每次40 ml,合并正丁醇提取液,减压浓缩至干,残渣加甲醇5 ml 溶解,作为对照药材溶液。另取缺淫羊藿的阴性样品适量,按供试品溶液制备方法,制得阴性对照溶液。照薄层色谱法(2010 年版《中国药典》(一部)附录ⅥB)试验[3],吸取上述供试品溶液3 μl、对照药材溶液和对照品溶液各2 μl、阴性对照溶液3 μl分别点于同一硅胶G薄层板上,以乙酸乙酯-丁酮-甲酸-水(10∶1∶1∶1,V/V/V/V)为展开剂,展开,取出,晾干,置于紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同颜色的荧光斑点。喷以10%硫酸乙醇溶液,于105 ℃加热至斑点显色清晰,置于日光下检视。结果,供试品色谱中,在与对照药材和对照品色谱相应的位置,显相同颜色的斑点。薄层色谱图见图1。

图1 淫羊藿薄层色谱鉴别色谱图A.365 nm荧光显色;B.10%硫酸乙醇溶液显色;1-3.供试品溶液(批号:130806、130807、130808);4.对照药材溶液;5.对照品溶液;6.阴性对照溶液Fig 1 TLC chromatogram of Epimedii FoliumA.TLC of 365 nm fluorescence;B.TLC of 10%H2SO4ethanol solution;1-3.test sample solution(sBatch No.130806,No.130807 and No.130808);4.reference medicinal materials;5.reference substance solution;6.negative control solution
2.1.2 黄芪 取3 批样品溶液各40 ml,分别加40%的氢氧化钠溶液5 ml,摇匀,用水饱和正丁醇提取3次,每次30 ml,合并正丁醇提取液,减压浓缩至干,残渣加甲醇2 ml使溶解,滤过,取滤液,作为供试品溶液。取黄芪甲苷对照品适量,加甲醇制成每1 ml含1 mg黄芪甲苷的溶液,作为对照品溶液。另取黄芪对照药材10 g,加甲醇50 ml,超声处理30 min,滤过,滤液用水饱和正丁醇提取2 次,每次40 ml,合并正丁醇提取液,减压浓缩至干,残渣加甲醇5 ml使溶解,作为对照药材溶液。另取缺黄芪的阴性样品适量,按供试品溶液制备方法,制得阴性对照溶液。照薄层色谱法(2010 年版《中国药典》(一部)附录ⅥB)试验[3],吸取上述供试品溶液3 μl、对照药材溶液和对照品溶液各2 μl、阴性对照溶液3 μl 分别点于同一硅胶G 薄层板上,以氯仿-乙酸乙酯-甲醇-水(20∶40∶22∶10,V/V/V/V)的下层溶液(10 ℃以下分层)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105 ℃加热5 min,置于日光下检视。结果,供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同的棕褐色斑点。薄层色谱图见图2。
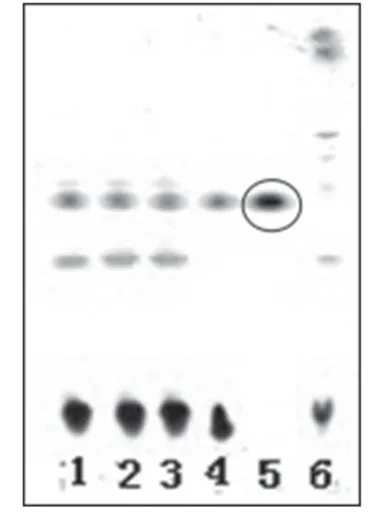
图2 黄芪薄层色谱鉴别色谱图1-3.供试品溶液(批号:130806、130807、130808);4.对照药材溶液;5.对照品溶液;6.阴性对照溶液Fig 2 TLC chromatogram of Astragali Radix1-3.test sample solution(sBatch No.130806,No.130807 and No.130808);4.reference medicinal materials;5.reference substance solution;6.negative control solution
2.1.3 枸杞 取3批样品溶液各50 ml,分别用乙酸乙酯提取3次,每次30 ml,合并乙酸乙酯提取液,减压蒸干,残渣加2 ml甲醇使溶解,作为供试品溶液。另取枸杞对照药材9 g,加水40 ml,加热煮沸15 min,放冷,滤过,滤液用乙酸乙酯提取2次,每次40 ml,合并乙酸乙酯提取液,减压浓缩至5 ml,作为对照药材溶液。另取缺枸杞子的阴性样品适量,按供试品溶液制备方法,制得阴性对照溶液。照薄层色谱法(2010 年版《中国药典》(一部)附录ⅥB)试验[3],吸取上述供试品溶液10 μl、对照药材溶液5 μl、阴性对照溶液10 μl 分别点于同一硅胶G薄层板上,以乙酸乙酯-氯仿-甲酸(3∶2∶1,V/V/V)为展开剂,展开,取出,晾干,置于紫外光灯(365 nm)下检视。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同颜色的荧光斑点。薄层色谱图见图3。
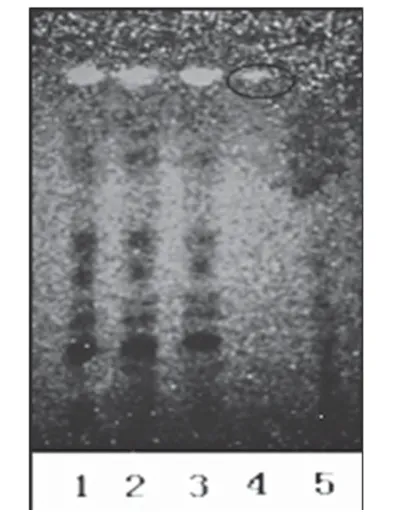
图3 枸杞薄层色谱鉴别色谱图1-3.供试品溶液(批号:130806、130807、130808);4.对照药材溶液;5.阴性对照溶液Fig 3 TLC chromatogram of lycium barbarum1-3.test sample solution(sBatch No.130806,No.130807 and No.130808);4.reference medicinal materials;5.negative control solution
2.1.4 白术 取3批样品溶液各50 ml,分别用乙酸乙酯提取3次,每次30 ml,合并乙酸乙酯提取液,减压蒸干,残渣加三氯甲烷2 ml使溶解,作为供试品溶液。另取白术对照药材1 g,加乙酸乙酯30 ml,超声处理15 min,滤过,滤液蒸干,同法制成对照药材溶液。另取缺白术的阴性样品适量,按供试品溶液制备方法,制得阴性对照溶液。照薄层色谱法(2010 年版《中国药典》(一部)附录ⅥB)试验[3],吸取上述供试品溶液10 μl、对照药材溶液5 μl、阴性对照溶液10 μl 分别点于同一硅胶G 薄层板上,以石油醚(60~90 ℃)-乙酸乙酯(50∶1,V/V)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热5 min,置于日光下检视。结果,供试品色谱中,在与对照药材色谱相应的位置上,显相同的红色斑点。薄层色谱图见图4。
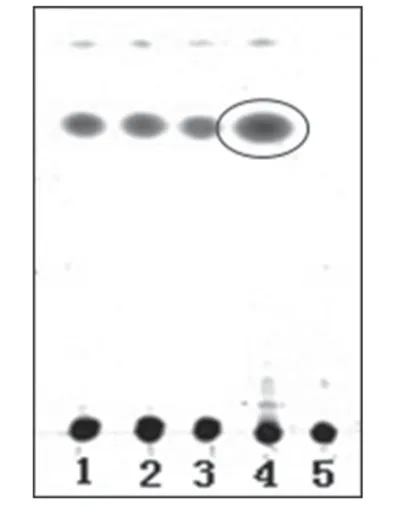
图4 白术薄层色谱鉴别色谱图1-3.供试品溶液(批号:130806、130807、130808);4.对照药材溶液;5.阴性对照溶液Fig 4 TLC chromatogram of Atracty lodes.macrocephala1-3.test sample solutions(Batch No.130806,No.130807 and No.130808);4.reference medicinal materials;5.negative control solution
2.1.5 人参 取“2.1.1”项下制得的供试品溶液,作为供试品溶液。取人参皂苷Rg1对照品适量,加甲醇制成每1 ml 升含1 mg 人参皂苷Rg1的溶液,作为对照品溶液。另取人参对照药材10 g,加甲醇50 ml,超声处理30 min,滤过,滤液用水饱和正丁醇提取2次,每次30 ml,合并正丁醇提取液,减压浓缩至干,残渣加甲醇5 ml 使溶解,作为对照药材溶液。另取缺人参的阴性样品适量,按供试品溶液制备方法,制得阴性对照溶液。照薄层色谱法(2010年版《中国药典》(一部)附录ⅥB)试验[3],吸取上述供试品溶液2 μl、对照药材溶液和对照品溶液各1 μl、阴性对照溶液2 μl 分别点于同一硅胶G 薄层板上,以氯仿-甲醇-水(13∶7∶2,V/V/V)的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105 ℃加热至斑点显色清晰,置于日光下检视。结果,供试品色谱中,在与对照药材和对照品色谱相应的位置,显相同颜色的斑点。薄层色谱图见图5。
2.2 含量测定
2.2.1 色谱条件 色谱柱:Dikma Diamonsil C18(250 mm×4.6 mm,5 μm);流动相:乙腈-0.1%磷酸水溶液(30∶70,V/V),等度洗脱;流速:1.0 ml/min;检测波长:270 nm;柱温:30 ℃;进样量:10 μl。
2.2.2 对照品溶液的制备 精密称取淫羊藿苷对照品1.02 mg,置于1 ml量瓶中,加甲醇溶解并稀释至刻度,即得对照品溶液(1.02 mg/ml)。
2.2.3 供试品溶液的制备 取本品10 ml,置于50 ml量瓶中,精密加入50%乙醇40 ml,密塞,超声处理30 min 后,放冷,再用50%乙醇定容,摇匀,滤过,取续滤液,即得。
2.2.4 阴性对照溶液的制备 除淫羊藿外,其他各药材按照仙人菇口服液的制备方法进行提取浓缩制成阴性对照样品后,按“2.2.3”项下方法制备,即得。

图5 人参薄层色谱鉴别色谱图1-3.供试品溶液(批号:130806、130807、130808);4.对照药材溶液;5.对照品溶液;6.阴性对照溶液Fig 5 TLC chromatogram of Panax.ginseng1-3.test sample solution(sBatch No.130806,No.130807 and No.130808);4.reference medicinal materials;5.reference substance solution;6.negative control solution
2.2.5 专属性试验 取上述对照品溶液、供试品溶液、阴性对照溶液各适量,按“2.2.1”项下色谱条件测定。结果,阴性对照溶液色谱中在淫羊藿苷色谱峰保留时间一致的位置上无色谱峰,表明其他药材对淫羊藿苷的含量测定无影响,详见图6。
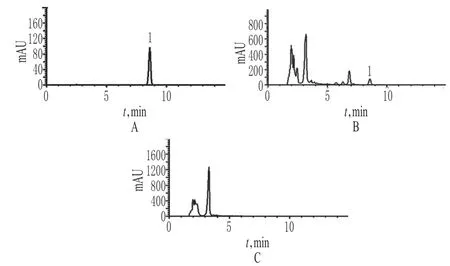
图6 高效液相色谱图A.对照品溶液;B.供试品溶液(批号:130806);C.阴性对照溶液;1.淫羊藿苷Fig 6 HPLC chromatogramsA.reference solution;B.test sample solution(Batch No.130806);C.negative control solution;1.icariin
2.2.6 线性关系考察 取“2.2.2”项下淫羊藿苷对照品溶液适量,加入甲醇稀释制成每1 ml 含510.00、255.00、127.50、63.75、31.88、15.94、7.97 μg的系列溶液,按“2.2.1”项下色谱条件进样测定,记录峰面积。以质量浓度(x,μg/ml)为横坐标、峰面积(y)为纵坐标进行线性回归,得淫羊藿苷线性回归方程为y=20.436x-76.473(r=0.999 3)。结果表明,淫羊藿苷质量浓度在7.97~510.00µg/ml范围内与峰面积呈良好的线性关系。
2.2.7 精密度试验 按“2.2.2”项下方法制备质量浓度为510 μg/ml的对照品溶液,按“2.2.1”项下色谱条件重复进样6次,记录峰面积。结果,淫羊藿苷峰面积的RSD为0.83%(n=6),表明仪器精密度良好。
2.2.8 稳定性试验 取“2.2.3”项下供试品溶液(批号:130806)适量,分别于提取后0、5、10、15、20、24 h 时按“2.2.1”项下色谱条件进样测定,记录峰面积。结果,淫羊藿苷峰面积的RSD为0.55%(n=6),表明供试品溶液在24 h内稳定性良好。
2.2.9 重复性试验 取同一样品(批号:130806)适量,按“2.2.3”项下方法制备供试品溶液6份,按“2.2.1”项下色谱条件进样测定,记录峰面积。结果,淫羊藿苷平均含量为305.98 μg/ml,RSD为0.45%(n=6),表明该方法重复性良好。
2.2.10 加样回收率试验 取同一批号(批号:130806)已知含量的样品各适量,共6 份,分别精密加入与样品中淫羊藿苷含量相等的对照品溶液,按“2.2.1”项下色谱条件进样测定,计算加样回收率,结果见表1。
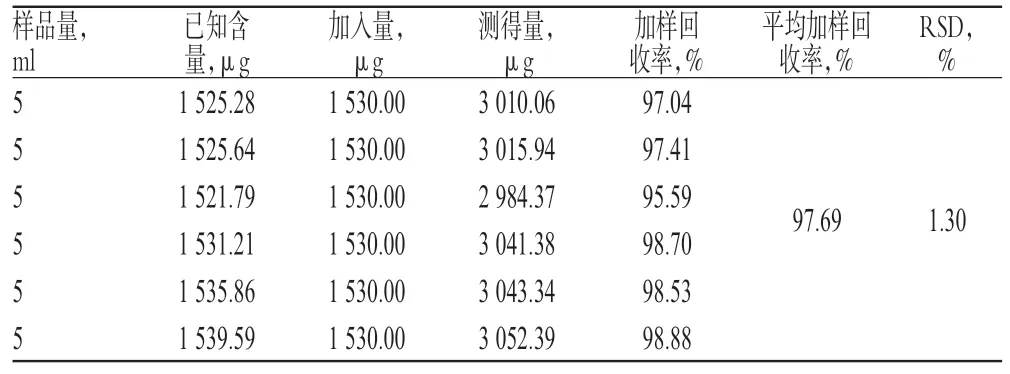
表1 加样回收率试验结果(n=6)Tab 1 Result of recovery tests(n=6)
2.2.11 样品含量测定 取3 批样品各适量,按“2.2.3”项下方法制备供试品溶液,再按“2.2.1”项下色谱条件进样测定并计算样品含量,结果见表2。

表2 样品含量测定结果(n=3)Tab 2 Result of content determination of samples(n=3)
3 讨论
3.1 TLC法
经比较,淫羊藿苷的薄层鉴别中喷以10%硫酸乙醇溶液显色也可见清晰斑点,且效果比《中国药典》中置于紫外光灯(365 nm)下检视所得荧光斑点更好。另外,TLC 中使用的显色剂中水的含量对斑点颜色和形状影响很大,无水乙醇和硫酸放置过久,含水量增加,显色剂显色不明显;且试验中易受边缘效应干扰而使样品斑点比移值(Rf)和形状异常变化。因此,严格控制展开剂中水的比例且现配现用,将展开缸在展开前进行预饱和,并将薄层板置于展开缸中间,可显著改善斑点的形状和颜色,减小边缘效应,使斑点清晰可见。
3.2 对照品纯度检查方法
采用二极管阵列检测器检测,样品在190~400 nm波长范围内,除样品峰外未检测到其他峰,样品峰峰面积百分比达到100%;且增加进样量至样品出现平头峰时,仍未检测出其他色谱峰。样品峰的峰纯度分析报告显示,峰纯度达到98%,理论板数为13 463。
3.3 色谱条件的选择
3.3.1 色谱柱的选择 本试验对3种型号的C18柱进行了考察(Dikma Diamonsil C18、Sapphire C18、Agilent Eclipse XDB-C18)。结果表明,三者均能较好地分离淫羊藿苷且峰形较好,出于成本考虑,选用Dikma Diamonsil C18。
3.3.2 柱温的选择 笔者考察了不同柱温(25、30、35 ℃)对分离效果的影响。结果表明,30 ℃条件下的分离效果略优,故选用30 ℃为本研究的柱温。
3.3.3 流动相的选择 本试验按照2010 年版《中国药典》(一部)[3]并结合其他相关文献[4-15]考察淫羊藿苷的流动相条件,分别对乙腈-水、乙腈-0.1%醋酸、乙腈-0.05%磷酸、乙腈-0.1%磷酸、乙腈-0.2%磷酸5种流动相系统进行测定试验。结果表明,乙腈-水为流动相,采用等度洗脱时淫羊藿苷出峰时间较晚,且色谱峰的对称性不好;加入醋酸或磷酸的流动相系统可提高淫羊藿苷出峰时间,但0.1%醋酸和0.05%磷酸的分离度较差,0.1%和0.2%磷酸条件下分离效果均较好。考虑到高浓度的磷酸会对色谱柱造成一定损伤,故选择乙腈-0.1%磷酸(30∶70,V/V)作为流动相,并且每次分析结束时加强对色谱柱和液相系统的冲洗和维护。
3.3.4 检测波长的选择 取淫羊藿苷对照品溶液适量,在190~400 nm波长范围内扫描,发现在270 nm波长处有最大吸收。参照2010年版《中国药典》(一部)[3]淫羊藿苷的检测波长,并结合其他相关文献[4-15],最终选择270 nm为检测波长。
3.4 供试品溶液制备方法的优化
3.4.1 提取方法的选择 精密量取10 ml 仙人菇口服液3 份,分别进行以下处理。①萃取法:加乙酸乙酯振摇提取3 次,每次20 ml,合并乙酸乙酯液,蒸干,残渣加甲醇50 ml溶解;②超声法:置于50 ml量瓶中,加甲醇40 ml,超声处理30 min,放冷,加甲醇至刻度,摇匀,滤过,取续滤液;③直接进样法:置于50 ml量瓶中,加甲醇至刻度,摇匀,滤过,取续滤液。使上述各样品取样量一致,测定峰面积,计算样品含量。结果,3种方法的质量浓度分别为262.99、309.01、300.35 μg/ml,表明超声法提取效果略优于萃取法和直接进样法,故选用超声法。
3.4.2 提取溶剂的选择 精密量取10 ml 仙人菇口服液4 份,分别加入不同溶剂(50%甲醇、100%甲醇、50%乙醇、95%乙醇)进行超声提取。使上述各样品取样量一致,测定峰面积,计算样品含量。结果,质量浓度分别为252.68、267.36、279.23、271.88 μg/ml,表明用50%乙醇作为提取溶剂时的提取效果较好,故选用50%乙醇作为提取溶剂。
3.4.3 提取药液比的选择 精密量取10 ml 仙人菇口服液3份,分别采用不同提取药液比(1∶2、1∶4、1∶10,V/V)进行超声提取。使上述各样品取样量一致,测定峰面积,计算样品含量。结果,质量浓度分别为179.69、285.51、274.77 μg/ml,表明当提取药液比为1∶4 时样品中淫羊藿苷含量最高,提取效果最好,故采用提取药液比为1∶4。
3.4.4 提取时间的选择 精密量取10 ml 仙人菇口服液3 份,分别采用不同超声时间(15、30、60 min)进行超声提取。使上述各样品取样量一致,测定峰面积,计算样品含量。结果,质量浓度分别为265.89、272.37、270.14 μg/ml,表明超声30 min时提取效果略优于其他时间,故采用超声提取30 min。
综上所述,该法快速、准确、操作简便,可作为仙人菇口服液的质量控制标准。
[1]施俊,许玲,秦志丰,等.仙人菇口服液治疗中晚期胃癌临床疗效观察[J].成都中医药大学学报,2002,25(1):15.
[2]陈天池,张霄峰,秦志丰,等.仙人菇口服液对59 例恶性肿瘤患者化疗中增效减毒作用的观察[J].中医杂志,2007,48(3):247.
[3]国家药典委员会.中华人民共和国药典:一部[S].2010年版.北京:中国医药科技出版社,2010:307、627、716、1 019、1 282.
[4]张柳,何静文.仙茸壮阳口服液中淫羊藿苷的含量测定[J].中国药业,2004,13(11):36.
[5]陈林,陈鸿平,刘友平.抗氧化口服液质量标准研究[J].时珍国医国药,2005,16(2):125.
[6]叶静,包国荣,曾森.康欣口服液质量标准的改进[J].时珍国医国药,2006,17(10):125.
[7]陈利,李卿.高效液相色谱法测定健汝美口服液中淫羊藿苷含量[J].中国药业,2006,15(18):26.
[8]范宁,刘春凤,王建明.RP-HPLC法测定还春口服液中淫羊藿苷的含量[J].中医药学报,2010,38(3):87.
[9]黄评贵,喻定开,孙树山,等.HPLC测定抗骨增生口服液淫羊藿苷含量[J].中成药,2003,25(9):774.
[10]张桂燕,刘斌,陈广耀.HPLC 测定复方芪藿口服液中淫羊藿苷的含量[J].中国中药杂志,2005,30(4):310.
[11]涂禾,马家骅,郭宏彦,等.HPLC法测定肾王膏中淫羊藿苷的含量[J].中国药房,2014,25(31):2 945.
[12]叶强,王燕.心通口服液中淫羊藿苷的含量测定[J].医药导报,2004,23(9):678.
[13]李萍.肾气归口服液质量标准的研究[J].现代医药卫生,2009,25(4):508.
[14]刘俊娥,孔筠.高效液相色谱法测定天蛾补肾口服液中淫羊藿苷的含量[J].山西医科大学学报,2010,41(12):1 045.
[15]巩伟,赵豫,赵庆华,等.十一味参龙口服液的质量标准研究[J].中国药房,2014,25(35):3 323.
猜你喜欢
食品与发酵工业(2023年21期)2023-11-26 07:50:24
中成药(2018年12期)2018-12-29 12:26:00
中成药(2018年1期)2018-02-02 07:20:01
中成药(2017年8期)2017-11-22 03:19:40
中成药(2017年4期)2017-05-17 06:09:49
中成药(2017年3期)2017-05-17 06:09:14
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:41
海峡科技与产业(2016年3期)2016-05-17 04:32:14
云南中医学院学报(2015年3期)2015-07-31 18:09:28
中国药业(2014年24期)2014-05-26 09:00:07
