
突变型p53与其合成致死基因的研究进展
2015-02-04 06:30刘同阳郭海强朱美妍黄英泽贾舒婷罗瑛张继虹
遗传 2015年4期
刘同阳,郭海强,朱美妍,黄英泽,贾舒婷,罗瑛,张继虹
突变型与其合成致死基因的研究进展
刘同阳1,2,郭海强2,朱美妍2,黄英泽2,贾舒婷2,罗瑛2,张继虹2
1. 昆明理工大学生命科学与技术学院,昆明 650500;2. 昆明理工大学医学院,衰老与肿瘤分子遗传学实验室,昆明 650500
恶性肿瘤的靶向治疗已经成为现阶段肿瘤治疗的热点。随着人们对癌基因认知的加深,借助合成致死的方法靶向治疗肿瘤已成为针对肿瘤特异性治疗的新策略。基因突变在肿瘤的形成和发展过程中具有重要作用。因此,了解肿瘤中与突变型基因有合成致死关系的靶基因的作用方式,有助于指导由突变型基因诱发肿瘤的个性化治疗。与突变型基因具有合成致死关系的靶基因可分为细胞周期调控基因和细胞非周期调控基因,文章综述了这两类靶基因与突变型基因如何构成合成致死作用以及此作用的现实意义。
;合成致死;合成致死的交互作用;肿瘤
现阶段肿瘤分子治疗技术除了沿用传统的细胞毒性作用进行治疗外,肿瘤的靶向治疗取得了很大的进步。目前临床上所使用的大部分化疗药物不仅能杀死快速增殖的癌细胞,同时也能杀死部分正常细胞,对人体产生了极大的毒副作用。因此,寻找癌症细胞与正常细胞的异常是提高抗癌药物特异性治疗和减小毒副作用的关键,识别肿瘤细胞中所特有的治疗靶点有助于提高治疗指数。
随着人们对肿瘤靶向治疗认识的不断提高,药物分子靶向治疗在治疗恶性肿瘤上得到了发展。在肿瘤靶向治疗的发展进程中,针对恶性肿瘤表型基因的靶向合成致死研究,成为探索和开发抗癌药物的新方法[1]。起初,合成致死(Synthetic lethality, SL)是指细胞中同时存在两个或者多个非致死的非等位基因突变时,导致细胞死亡的现象[2]。现在,合成致死的意义更多地被用于肿瘤治疗,指肿瘤中致癌基因与其靶基因共同作用杀伤肿瘤。当肿瘤细胞中的致癌基因与对应的靶点基因存在合成致死关系时,能够抑制或激活靶基因而导致细胞死亡的相互作用称之为合成致死的交互作用(Synthetic lethal interactions)。合成致死的交互作用泛指借助治疗药物与致癌基因协同作用杀伤肿瘤细胞[3,4](图1)。聚腺苷二磷酸核糖聚合酶(Poly-ADP-ribose polymerase, PARP)抑制剂治疗BRCA(Breast and ovarian cancer susceptibility gene, BRCA)突变的乳腺癌正是利用了合成致死的原理[5,6]。因此,寻找与致癌基因具有合成杀伤作用的靶基因,发挥合成致死作用可以作为肿瘤靶向治疗的新方向。
基因在肿瘤中突变率较高,超过50%的肿瘤中均存在基因的突变,因此研究与突变型基因相关的合成致死关系,探索与肿瘤中突变型基因相关的靶向合成致死作用,将成为未来靶向抗癌药物研究的重要发展方向。
1 p53与肿瘤
于1979年首次被发现,最初人们认为是一个原癌基因。随着对的深入探究,研究人员发现在不同肿瘤中存在突变型(Mutant, mut)和野生型(Wild type, wt)两种形态[7~9]。Donehower等[10]发现敲除的小鼠具有高致瘤性,后来证实wt在体内有明确的抑癌作用,从此被更正为抑癌基因。作为转录因子,主要通过转录调节下游的靶基因发挥肿瘤抑制作用。在DNA损伤、原癌基因活化、缺氧和微管损伤等压力条件下,在信号转导过程中活化。活化后的具有调节细胞周期阻滞、诱导细胞凋亡、维持基因组稳定、诱导衰老和抑制血管生成等作用,阻止了DNA损伤和有丝分裂后异常染色体分布细胞的存活,从而发挥的保护效应(调节周期阻滞进行自我修复或者促使细胞程序性死亡)[11]。目前,成年人中超过50%的恶性肿瘤中都伴随有基因的突变。研究表明,mut丧失了wt的肿瘤抑制功能,突变后会发生功能获得(Gain of function),而获取了新的癌基因活性,如促进肿瘤细胞的增殖、存活、代谢、血管生成和转移,并且抑制了家族、的活性等[12,13]
2 p53与合成致死
近年来,关于mut与其存在合成致死关系基因的报道越来越多。在各类肿瘤中mut与其对应的靶基因存在着复杂的合成致死关系。综合对合成致死的种种研究成果,可从周期调控基因和非周期调控基因两方面来阐述其相应的合成致死关系。本文从周期调控相关基因以及非周期调控基因两个角度,阐述与mut存在合成致死关系靶基因的作用关系。
2.1 周期调控相关基因与mut p53存在的致死关系
作为“基因卫士”参与细胞周期调控。在细胞周期中,的调节功能主要表现为参与G1和G2/M 期检验点的检测。当细胞出现DNA损伤时,得以活化。此时,可以激活下游相关基因的表达,使细胞周期阻滞在G1和G2/M期阶段并进行DNA损伤修复,这就是细胞周期G1和G2/M期检验点[14]。与此同时,若产生的DNA损伤不能被机体修复,可反式激活和等凋亡基因来阻止细胞中DNA损伤的积累。Mut失去了介导的周期阻滞和DNA修复的功能,导致遗传信息错误的细胞越过周期检验点检测无限制增值,最终形成恶性肿瘤。
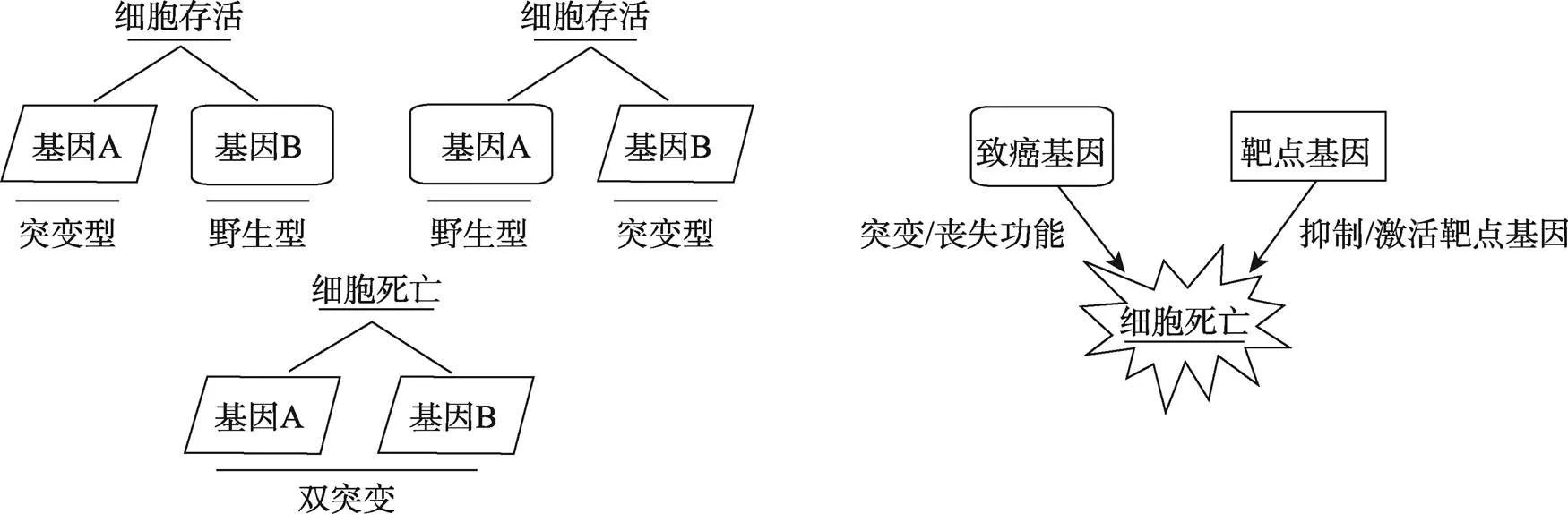
图1 合成致死与合成致死的交互作用
在wt细胞中出现DNA损伤时,G1和G2/M细胞周期检验点活化,阻止了DNA损伤程度的积累, DNA损伤修复通过作用G1和G2/M细胞周期检验点来进行细胞周期阻滞和修复。在mut的肿瘤细胞中,由于缺乏wt调控的细胞G1期检验点,所以当细胞的DNA遭受损伤时更依赖于G2/M细胞周期检验点。有文献报道[15]使用UCN-01(小分子化合物,细胞G2/M细胞周期检验点阻断剂)对mut肿瘤治疗起到化疗增敏的作用,研究结果表明mut细胞受化疗损伤时,阻断G2/M细胞周期检验点可以诱发凋亡。然而在mut细胞中,DNA损伤修复受WEE1(属核丝氨酸/苏氨酸蛋白激酶家族成员)的调控。WEE1在mut细胞的G2/M细胞周期检验点中执行重要作用,可以介导G2/M细胞周期检验点的活化、抑制周期蛋白依赖性激酶1(Cyclin-dependent kinase 1,CDK1)磷酸化,阻止肿瘤细胞周期进程[16]。已有大量文献报道[17-20]在mut的细胞中出现DNA损伤时,抑制WEE1功能可以杀伤mut的肿瘤细胞,产生抗肿瘤效应。因此WEE-1抑制剂与mut存在合成致死作用,寻找WEE-1抑制剂可以作为未来针对mut肿瘤细胞治疗药物的开发方向。
在G2/M细胞周期检验点中,其他相关靶点分子也与mut存在合成致死关系,如检验点激酶1(Checkpoint kinase 1,CHK1)、双特异性激酶 (Dual- specificity kinase,MYT1)和共济失调毛细血管扩张突变 (Ataxia telangiectasia mutated, ATM)等。研究证明,在G2/M期检验点调节机制中,如CHK1、MYT1和ATM等在mut细胞中敲除或受到抑制后也会对mut细胞系显示出杀伤性[18,21~23]。因此G2/M细胞周期检验点相关调控激酶——CHK1、MYT1和ATM与mut有合成致死作用。
除此之外,在DNA损伤时,抑制MAPK激活蛋白激酶2(MAP-kinase activated protein kinase 2, MK2)的功能,可以对mut细胞产生杀伤作用[24]。在mut的细胞中,细胞有丝分裂依赖ATM和ATR介导的细胞周期检验点信号通路,在DNA损伤后,细胞通过p38 MAPK/MK2途径存活。当MK2缺失后,双重特异性磷酸酶Cdc 25A/B的磷酸化水平大大降低,导致此类肿瘤细胞有丝分裂障碍,从而导致肿瘤细胞的死亡。因此,MK2在mut的细胞中对细胞周期检验点的功能起着重要的作用,研究结果证实了在此通路中通过沉默MK2可以杀伤存在mut的肿瘤细胞[25,26]。
研究报道[27],丝氨酸/苏氨酸蛋白激酶Polo样激酶1(Polo-like kinase 1,PLK1)为G2/M期检验点调节剂,丧失wt功能的肿瘤细胞生存依赖于PLK1,当机体内丧失wt功能时PLK1起着重要的细胞周期调节作用。科研人员通过体内、体外研究发现[28,29],mut和wt基因功能缺失()细胞系对PLK1抑制剂具有很强的敏感性。PLK1抑制剂能够作为一种单一因素引发和mut细胞的凋亡,故PLK1抑制剂可以对mut和基因功能缺失的肿瘤细胞系产生合成致死作用。因此,PLK1也可以设计成针对突变和基因功能缺失合成致死的靶点。综上所述,细胞周期检验中mut介导的合成致死如图2所示。
2.2 与mut p53产生合成致死交互作用的其他因素
最初美国国家癌症研究所(NCI)对60种mut的细胞系进行了多种化合物的筛选,旨在选出活性较好的先导化合物作为治疗mut细胞系的备选抗癌药物。研究者共筛选出包括紫杉醇在内对mut肿瘤细胞有较好的杀伤性作用的多个化合物。随后Zhang等[30]在对紫杉醇的深入研究中发现,紫杉醇处理mut肿瘤细胞后影响了微管相关蛋白-4(Microtubule-associated protein 4, MAP-4)的表达,MAP-4可以诱导细胞在有丝分裂前所需微管的形成。紫杉醇在mut肿瘤细胞中稳定并促进了MAP-4诱导微管的表达,具体表现为紫杉醇处理mut的肿瘤细胞后会产生和积累大量的微管,这些微管的积累影响了肿瘤细胞的多种功能,特别是会使细胞分裂停止于有丝分裂期,阻断了细胞的正常有丝分裂进程,从而导致细胞死亡。随后Meng等[31]在紫杉醇对mut的子宫内膜癌研究中进一步发现,抗血管活性化合物BIBF1120(VEGFR、PDGFR、FGFR酪氨酸激酶抑制剂)可以在mut的细胞系中稳定紫杉醇的功能发挥,调节紫杉醇耐药性问题。研究证实,血管激酶抑制剂BIBF1120可以被用来恢复紫杉醇处理mut子宫内膜癌细胞有丝分裂的敏感性。综上所述,紫杉醇通过作用于MAP-4对mut肿瘤细胞起到杀伤作用,所以MAP-4可以作为一个设计mut合成致死关系的靶点。

图2 细胞周期检验中mut p53介导的合成致死
在mut p53功能的肿瘤细胞中,CHK1、WEE1、MK2、ATM、PLK1和 MTY1通过G2/M细胞周期检验途径与mut p53产生合成致死交互作用,其中CHK1、WEE1、MK2和ATM在出现DNA损伤时与mut p53存在合成致死关系。
在人类癌症中,基因的R175H位点突变是一个最常见的突变。Hiroo等[32]在近期研究中发现,使用siRNA干扰分化抑制因子1(Inhibitor of differentiation 1, ID1)会抑制R175H位点突变的细胞的生长,但是在-null和R273H的细胞中却未发现此现象,由此显示ID1与p53存在合成致死关系。ID1抑制剂作用于p53位点突变细胞系时会产生合成致死的交互作用。
糖尿病治疗药物二甲双胍可以杀伤mut的细胞[33],二甲双胍可以激活腺苷酸活化蛋白激酶(AMP activated protein kinase, AMPK)代谢体内的糖分,然而在mut的肿瘤细胞中主要依赖于糖酵解途径产生能量,二甲双胍可以竞争mut肿瘤中的糖分,剥夺其能量的产生,从而诱导凋亡的发生。AMPK活化可形成一个使mut细胞更易受损的环境。AMPK与mut之间也有着重要的关联,AMPK激动剂有望成为与mut具有合成致死作用的靶分子。
在mut的细胞中,mut与鸟苷酸交换因子H1(Guanine nucleotide exchange factor-H1, GEF-H1)的表达存在协同关系,在mut的肿瘤细胞系中GEF-H1的表达随着mut表达的增加而增加。在mut的肿瘤细胞中GEF-H1参与的能量转换过程为肿瘤的形成提供能量支持,从而促进mut细胞的增殖。现已证实mut的细胞生长依赖于GEF-H1,抑制GEF-H1可以抑制mut细胞的生长。因此GEF-H1与mut存在合成致死关系,GEF-H1有望成为一个与mut相联系的合成致死靶点[34]。
Baldwin等[35]使用HPV(人类乳头瘤病毒)感染的宫颈细胞系构建mut细胞系,利用shRNA干扰技术靶向一些与关联的基因,发现在丝氨酸/苏氨酸蛋白激酶中,血清糖皮质激素诱导激酶2(Serum and Glucocorticoid Induced Kinase 2, SGK2)和活化激酶3(P21 protein -activated kinase 3, PAK3)与mut之间有紧密的关联作用。研究结果表明,SGK2、PAK3的表达会影响mut细胞的存活,但是两者对于mut肿瘤细胞的影响具有不同的作用机制。在mut的细胞中,敲低SGK2的表达时会导致细胞自噬,抑制PAK3的表达时会导致细胞凋亡蛋白酶caspase-3激活,从而诱发细胞凋亡。这些研究表明,SGK2和PAK3的抑制剂可以选择性杀死mut的肿瘤细胞,两者均在mut的细胞中展现出杀伤作用,因此,SGK2和PAK3的抑制剂对于mut存在合成致死作用。综上所述,相对应的合成致死关系如图3所示。

图3 非周期激酶与p53的合成致死关系
MAP-4、PAK3、GEF-H1、AMPK、SGK2、ID1和mut存在合成致死关系。作用于MAP-4、PAK3、GEF-H1、AMPK、SGK2和ID1均与mut存在合成致死作用,MAP-4与mut的合成致死关系是通过诱导微管异常导致细胞死亡,PAK3抑制剂与mut诱发的合成致死作用通过诱导凋亡途径。抑制GEF-H1与激活AMPK通过参与mut肿瘤的能量转换过程,从而诱导了凋亡的发生。抑制SGK2和ID1与mut产生的合成致死作用的途径尚未明确。
3 mut p53的合成致死研究在肿瘤治疗中的应用前景
致癌基因的靶向作用治疗已经成为肿瘤治疗的新兴领域。越来越多的研究证明了恶性肿瘤靶向治疗具有较好的高效性和特异性。甲磺酸伊马替尼针对融合基因治疗的成功出现,在分子癌症治疗中具有里程碑的意义。肿瘤多数由基因突变引起,基因突变的多样性成为肿瘤治疗的难点。在肿瘤治疗中运用合成致死的方法有助于了解致癌基因与其相关基因、致癌基因与药物靶分子之间的作用关系,进一步制定肿瘤的靶基因特异性治疗策略和开发出针对致癌基因的特异性治疗药物,将对未来肿瘤治疗学研究具有现实的意义。
在人类恶性肿瘤中,抑癌基因作为高频突变基因是目前肿瘤治疗的首要靶基因。随着科研人员对mut的深入研究,mut与其所必需的关联基因调节生物学进程的作用关系逐渐被人们所发掘。针对与mut存在相关作用的靶基因,设计药物分子与肿瘤中mut产生合成致死作用而杀死肿瘤细胞,有望成为肿瘤治疗的方法之一。未来针对肿瘤中mut进行合成致死药物筛选,将会是合成致死作用筛选的首要任务。总之,寻找肿瘤中mut的合成致死靶基因,针对靶基因和mut开发相关合成致死药物将会成为未来肿瘤治疗史上具有重要意义的研究方向。
[1] Dörr JR, Yu Y, Milanovic M, Beuster G, Zasada C, Däbritz JH, Lisec J, Lenze D, Gerhardt A, Schleicher K, Kratzat S, Pürfurst B, Walenta S, Mueller-Klieser W, Gräler M, Hummel M, Keller U, Buck AK, Dörken B, Willmitzer L, Reimann M, Kempa S, Lee S, Schmitt CA. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy., 2013, 501(7467): 421–425.
[2] Chan DA, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery., 2011, 10(5): 351–364.
[3] Pessetto ZY, Yan Y, Bessho T, Natarajan A. Inhibition of BRCT(BRCA1)-phosphoprotein interaction enhances the cytotoxic effect of olaparib in breast cancer cells: a proof of concept study for synthetic lethal therapeutic option., 2012, 134(2): 511–517.
[4] Dietlein F, Thelen L, Jokic M, Jachimowicz RD, Ivan L, Knittel G, Leeser U, Van Oers J, Edelmann W, Heukamp LC, Reinhardt HC. A functional cancer genomics screen identifies a druggable synthetic lethal interaction betweenand., 2014, 4(5): 592–605.
[5] Warrener P, Kim S, Williams SM, Biery M, Gordon M, Toniatti C, Cleary MA, Linsley PS, Carleton M. Synthetic lethality of PARP inhibition in BRCA-network disrupted tumor cells is associated with interferon pathway activation and enhanced by interferon-γ., 2012, 17(7): 691–701.
[6] Chan SL, Mok T. PARP inhibition in BRCA-mutated breast and ovarian cancers., 2010, 376(9737): 211–213.
[7] Eliyahu D, Michalovitz D, Eliyahu S, Pinhasi-Kimhi O, Oren M. Wild-type p53 can inhibit oncogene-mediated focusformation., 1989, 86(22): 8763–8767.
[8] Steele RJ, Lane DP. P53 in cancer: a paradigm for modern management of cancer., 2005, 3(3): 197–205.
[9] Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P, Glover T, Collins FS, Weslon A, Modali R, Harris CC, Vogelstein B. Mutations in the p53 gene occur in diverse human tumour types., 1989, 342(6250): 705–708.
[10] Donehower LA, Harvey M, Slagle BL, Mcarthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours., 1992, 356(6366): 215–221.
[11] Vousden KH, Lu X. Live or let die: the cell's response to p53., 2002, 2(8): 594–604.
[12] Liu J, Zhang C, Feng Z. Tumor suppressor p53 and its gain-of-function mutants in cancer., 2014, 46(3): 170–179.
[13] Kastan MB, Berkovich E. p53: a two-faced cancer gene., 2007, 9(5): 489–491.
[14] Dasika GK, Lin SC, Zhao S, Sung P, Tomkinson A, Lee EY. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis., 1999, 18(55): 7883–7899.
[15] Wang Q, Fan S, Eastman A, Worland PJ, Sausville EA, O'connor PM. UCN-01: a potent abrogator of G2 checkpoint function in cancer cells with disrupted p53., 1996, 88(14): 956–965.
[16] Rowley R, Hudson J, Young PG. The wee1 protein kinase is required for radiation-induced mitotic delay., 1992, 356(6367): 353–355.
[17] Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, Itadani H, Takahashi-Suzuki I, Fukasawa K, Oki H, Nambu T, Jiang J, Sakai T, Arakawa H, Sakamoto T, Sagara T, Yoshizumi T, Mizuarai S, Kotani H. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents., 2009, 8(11): 2992–3000.
[18] Wang Y, Decker SJ, Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis., 2004, 3(3): 305–313.
[19] Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, Reigan P, Porter CC. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeuticsand, independent of p53 functionality., 2013, 12(12): 2675–2684.
[20] Pappano WN, Zhang Q, Tucker LA, Tse C, Wang J. Genetic inhibition of the atypical kinase Wee1 selectively drives apoptosis of p53 inactive tumor cells., 2014, 14: 430.
[21] Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response., 2009, 23(16): 1895–1909.
[22] Wang X, Ma Z, Xiao Z, Liu H, Dou Z, Feng X, Shi H. Chk1 knockdown confers radiosensitization in prostate cancer stem cells., 2012, 28(6): 2247–2254.
[23] Biddlestone-Thorpe L, Sajjad M, Rosenberg E, Beckta JM, Valerie NC, Tokarz M, Adams BR, Wagner AF, Khalil A, Gilfor D, Golding SE, Deb S, Temesi DG, Lau A, O'connor MJ, Choe KS, Parada LF, Lim SK, Mukhopadhyay ND, Valerie K. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation., 2013, 19(12): 3189–3200.
[24] Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53- deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage., 2007, 11(2): 175–189.
[25] Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T, Yaffe MB. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo., 2013, 5(4): 868–877.
[26] Razzak M. Basic research: MK2 and p53 - a lethal pairing., 2014, 11(1): 3.
[27] Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53., 2009, 106(10): 3964–3969.
[28] Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, Zhang X, Bleam M, Halsey W, Hughes A, Moy C, Liu-Sullivan N, Powers S, Bachman K, Jackson J, Weber B, Wooster R. Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability., 2010, 9(7): 2079–2089.
[29] Tyagi S, Bhui K, Singh R, Singh M, Raisuddin S, Shukla Y. Polo-like kinase1 (Plk1) knockdown enhances cisplatin chemosensitivity via up-regulation of p73α in p53 mutant human epidermoid squamous carcinoma cells., 2010, 80(9): 1326–1334.
[30] Zhang CC, Yang JM, Bash-Babula J, White E, Murphy M, Levine AJ, Hait WN. DNA damage increases sensitivity to vinca alkaloids and decreases sensitivity to taxanes through p53-dependent repression of microtubule-associated protein 4., 1999, 59(15): 3663–3670.
[31] Meng X, Dizon DS, Yang S, Wang X, Zhu D, Thiel KW, Leslie KK. Strategies for molecularly enhanced chemotherapy to achieve synthetic lethality in endometrial tumors with mutant p53., 2013, 2013: 828165.
[32] Imai H, Kato S, Sakamoto Y, Kakudo Y, Shimodaira H, Ishioka C. High throughput RNAi screening identifies ID1 as a synthetic sick/lethal gene interacting with the common TP53 mutation R175H., 2014, 31(3): 1043–1050.
[33] Buzzai M, Jones RG, Amaravadi RK, Lum JJ, Deberardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth., 2007, 67(14): 6745–6752.
[34] Mizuarai S, Yamanaka K, Kotani H. Mutant p53 induces the GEF-H1 oncogene, a guanine nucleotide exchange factor-H1 for RhoA, resulting in accelerated cell proliferation in tumor cells., 2006, 66(12): 6319–6326.
[35] Baldwin A, Grueneberg DA, Hellner K, Sawyer J, Grace M, Li W, Harlow E, Munger K. Kinase requirements in human cells: V. Synthetic lethal interactions between p53 and the protein kinases SGK2 and PAK3., 2010, 107(28): 12463–12468.
(责任编委: 吴晨)
Synthetic lethal genes to mutant
Tongyang Liu1,2, Haiqiang Guo2, Meiyan Zhu2, Yingze Huang2, Shuting Jia2, Ying Luo2, Jihong Zhang2
Targeted therapy has become a powerful approach for cancer treatment. Better understanding of oncogenes as well as synthetic lethal interactions with oncogenes will lead to new strategies for tumor-specific treatment. It is well known that mutantplays an important role in tumorigenesis and tumor development. Thus, understanding the synthetic lethal relationship betweenmutations and interacting genes in tumor is critical for the personalized treatments ofmutant tumors. Synthetic lethal genes to mutantcan be divided into cell cycle regulators and non-cell cycle regulators. This paper review show these two types of target genes contribute to synthetic lethal interactions withmutations and potential applications of these interactions in anticancer therapy.
; synthetic lethality; synthetic lethal interactions; tumor
2014-08-18;
2014-09-28
国家自然科学基金项目(编号:81260501,U1202221)资助
刘同阳,硕士研究生,专业方向:肿瘤药理学。E-mail: sunshine_tongyang@163.com
张继虹,博士,教授,研究方向:分子药理学。E-mail: zhjihong2000@126.com
10.16288/j.yczz.14-277
2014-12-8 11:46:34
http://www.cnki.net/kcms/detail/11.1913.R.20141208.1146.002.html
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2021年12期)2021-12-30
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中成药(2018年12期)2018-12-29
中国医药生物技术(2015年4期)2015-12-26
医学研究杂志(2015年4期)2015-06-10
中国医药导报(2015年26期)2015-02-28
中华介入放射学电子杂志(2014年1期)2014-02-02
中国医学科学院学报(2013年6期)2013-03-11
