
盐酸巴尼地平的合成工艺改进*
2015-02-02 07:53:41周梦月江传亮陈国华中国药科大学药物化学教研室江苏南京0009南京天海医药科技有限公司江苏南京0048
合成化学 2015年7期
周梦月,江传亮,陈国华(.中国药科大学药物化学教研室,江苏南京 0009; .南京天海医药科技有限公司,江苏南京 0048)
盐酸巴尼地平的合成工艺改进*
周梦月1,江传亮2,陈国华1
(1.中国药科大学药物化学教研室,江苏南京210009; 2.南京天海医药科技有限公司,江苏南京210048)
摘要:3-羟基丙腈与双乙烯酮反应后与间硝基苯甲醛和3-氨基巴豆酸甲酯经“一锅煮”反应制得中间体2,6-二甲基-4-(间硝基苯基) -1,4-二氢吡啶-5-羧酸甲酯-3-羧酸(4) ; 4用奎尼丁拆分得(R) -4; (R) -4与(S) -N-苄基-3-羟基吡咯烷经酯化和成盐反应合成了盐酸巴尼地平,总收率12.7%,其结构经1H NMR,IR和ESI-MS确证。
关键词:盐酸巴尼地平; 1,4-二氢吡啶;药物合成;工艺改进
盐酸巴尼地平[(3S) -1-苄基-3-吡咯烷基甲基-(4S) -2,6-二甲基-4-(间硝基苯基) -1,4-二氢吡啶-3,5-二羧酸酯盐酸盐(1),Chart 1]是日本山之内公司研制开发的新型长效二氢吡啶类钙离子拮抗剂。研究表明,1可以在不影响心率的前提下[1]通过特异性地作用细胞跨膜电位依赖性钙离子通道,抑制钙流入细胞内,选择性地使外周血管及冠状血管的平滑肌松弛从而达到降血压的作用。临床上1主要用于治疗原发性的高血压及肾性高血压[2]。

Chart 1
文献[3]对1的合成进行了详细的总结,路线有三条。路线一:以乙酰乙酸甲酯为起始原料[4]。该路线中合成的中间体对称双甲酯要经过单侧水解得到单羧酸酯,反应过程中易生成双水解副产物,产品纯度较低,对后续反应影响大;以L-(-) -苹果酸拆分巴尼地平,收率低(25.8%)。路线二:苄基吡咯烷醇和双乙烯酮反应后,再与间硝基苯甲醛,氨基巴豆酸甲酯反应得制得1[5]。该路线每步都需经柱层析纯化,不适合工业化生产。路线三:以叔丁基乙酰乙酸酯为起始原料,与间硝基苯甲醛反应得到关键中间体单羧酸酯,但该步反应收率低(32.7%),且需柱层析纯化,亦不利于工业化生产[6]。
本文综合以上三条路线的优缺点,选择了一条更为合理的路线合成1。并在文献[3,7-8]方法的基础上对1的合成工艺进行改进,旨在简化合成工艺的同时,提高1的合成总收率。以3-羟基丙腈(2)和双乙烯酮为原料,经加成反应得乙酰乙酸(2-氰基)乙酯(3) ; 3与间硝基苯甲醛和3-氨基巴豆酸甲酯反应,经选择性水解得到2,6-二甲基-4-(间硝基苯基) -1,4-二氢吡啶-5-羧酸甲酯-3-羧酸(4) ; 4用奎尼丁拆分得(R) -4; (R) -4再与由L-苹果酸和苄胺反应还原得到的(S) -N-苄基-3-羟基吡咯烷[(S) -6]酯化、成盐合成了1 (Scheme 1),总收率12.7%,其结构经1H NMR,IR和ESI-MS确证。
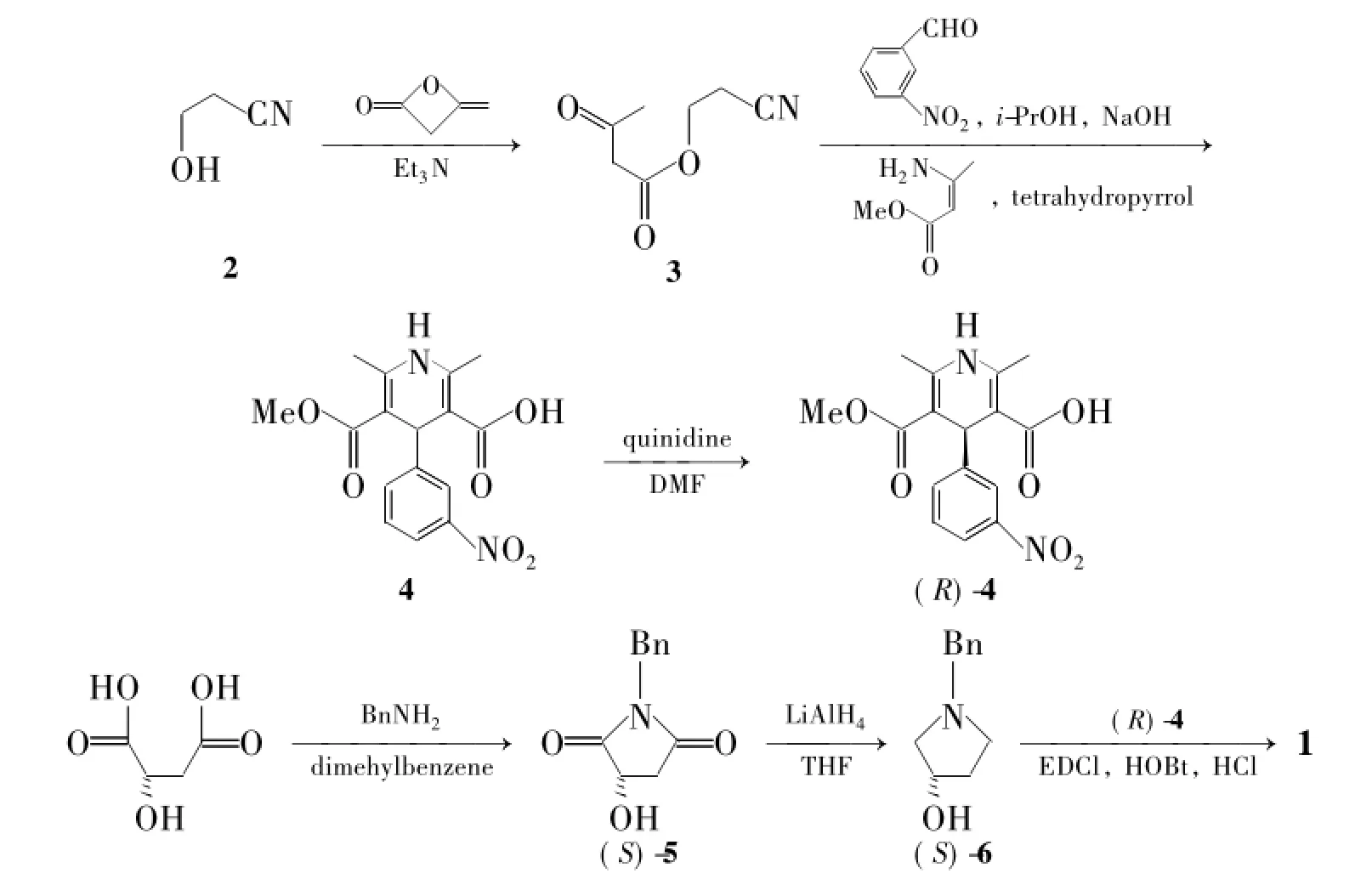
Scheme 1
1 实验部分
1.1仪器与试剂
RY-1型熔点仪(温度未校正) ; WZZ 2B型自动旋光仪; Bruker AV-300 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标) ; Nicolet Impact 410型红外光谱仪(KBr压片) ; Agilent 1100 ESI-MS型质谱仪; 10-AP型高效液相色谱仪。
异丙醇,丙酮,乙酸乙酯,DMF,THF,二氯甲烷、二甲苯和乙腈,工业级;间硝基苯甲醛,灌南伊斯特化工有限公司;其余所用试剂均为化学纯或分析纯。
1.2合成
(1) 3的合成
在干燥反应瓶中加入3-羟基丙腈58.5 g(824 mmol)和三乙胺2.0 g,搅拌下于80℃缓慢滴加双乙烯酮79.5 g(946 mmol),滴毕,于80℃反应6 h。旋蒸除去低沸点物质得红棕色油状液体3 79.2 g,收率62.0%;1H NMR δ: 2.29(s,3H),2.71~2.77(t,J =6.3 Hz,2H),3.54(s,2H),4.33~4.38(t,J =2.8 Hz,2H)。
(2) 4的合成
在反应瓶中依次加入3 20.1 g(130 mmol),间硝基苯甲醛15.1 g(100 mmol),催化剂四氢吡咯0.57 g和异丙醇150 mL,搅拌下于45℃反应3 h。于55℃缓慢加入3-氨基巴豆酸甲酯14.0 g (122 mmol),反应0.5 h。冷却至室温,控制温度(50℃)迅速减压蒸馏除去溶剂,残余物加丙酮100 mL,搅拌下于-8℃缓慢滴加10%氢氧化钠溶液60 mL,滴毕,反应1.5 h。减压浓缩,残余物加水80 mL,强力搅拌后过滤,滤液用乙酸乙酯(4×20 mL)洗涤,水层在冰浴冷却下用浓盐酸调至pH 6.0~6.5。过滤,滤饼用甲醇洗涤,干燥得淡黄色固体4 21.5 g,收率64.7% (56.3%[3]),m.p.202℃~204℃(199℃~202℃[4]),纯度92.5%[HPLC归一化法: SHIMADZU C18 ODS色谱柱(4.6 mm×150 mm,5 μm) ;流动相: V(pH 3.5磷酸溶液)∶V(甲醇) = 2∶3;检测波长: 236 nm;柱温: 25℃;流速: 1 mL·min-1,下同];1H NMR δ: 2.29(s,6H),3.56(s,3H),5.01 (s,1H),7.52~8.03 (m,4H),8.99 (br s,1H),11.90(s,1H)。
(3) (R) -(4)的合成
在反应瓶中依次加入4 5.0 g(15 mmol),奎尼丁4.9 g(15 mmol),DMF 24 mL和水15 mL,加热搅拌使其完全溶解;于室温静置24 h。过滤,滤饼干燥得淡黄色固体,加入3%氢氧化钠溶液10 mL,搅拌均匀得悬浊液,用二氯甲烷(3×30 mL)洗涤,水层用稀盐酸调至pH 6.4。过滤,滤饼干燥得淡黄色固体(R) -4 2.0 g,收率40.6%,m.p.201℃~203℃,[α]2D5-23.5°(c 5 mg· mL-1,丙酮) (26%,200℃~201℃,-21.9°[9])。
(4) (S) -5的合成
在反应瓶中加入L-(-)苹果酸13.1 g(100 mmol),搅拌下升温至110℃使其完全熔融;滴加苄胺10.2 g(100 mmol),滴毕,于170℃~175℃带水反应2 h。加二甲苯150 mL,于185℃~190℃(浴温)回流带水反应2 h。冷却至室温,析出大量固体,过滤,滤饼用甲苯洗涤,干燥得类白色固体(S) -5 14.2 g,收率67.1%,m.p.110℃~111℃,[α]2D5-48.57°(c 10 mg·mL-1,甲醇) (-51.1°[4]) ;1H NMR δ: 2.45~2.47(m,1H),3.02~3.11 (m,1H),4.59~4.61(d,J = 5.7 Hz,1H),4.55~4.58(s,2H),6.14~6.17(br s,1H),7.25~7.36(m,5H)。
(5) (S) -6的合成
在反应瓶中加入5 5.0 g(24 mmol)和无水THF 130 mL,冰浴冷却,搅拌下于10℃~20℃缓慢加入四氢锂铝2.8 g(72 mmol),加毕,于65℃反应4 h。冷却至室温,于<10℃(内温)依次缓慢滴加纯净水(2.8 mL),15%氢氧化钠溶液(2.8 mL)和纯净水(8.4 mL)。过滤,滤饼用乙酸乙酯(3×30 mL)洗涤,合并滤液和洗液,减压蒸除溶剂,残余物用乙酸乙酯(20 mL)溶解,用饱和食盐水(3×30 mL)洗涤,10%盐酸水溶液(3×30 mL)萃取,水层用15%氢氧化钠溶液调至pH约9,用乙酸乙酯(3×30 mL)萃取,合并有机层,用无水硫酸钠干燥过夜。减压蒸除溶剂得淡黄色油状液体(S) -6 3.5 g,收率87.5%,纯度95.5%,[α]2D5-2.92°(c 30 mg·mL-1,甲醇) (收率76%,-3.10°[4]) ;1H NMR δ: 1.13~1.21(m,1H),1.46~1.52(m,1H),1.90~2.20(m,2H),2.23~2.29(m,1H),2.31~2.39(m,1H),3.96(m,1H),4.15(s,2H),5.72~5.73 (br s,1H),7.43~7.58(m,5H)。
将(S) -6 3.5 g用15 mL乙酸乙酯溶解;通入氯化氢气体,析出白色固体。过滤,滤饼干燥得(S) -6·HCl。
(6) 1的合成
在反应瓶中加入(R) -4 3.3 g(10 mmol)和无水二氯甲烷15 mL,搅拌使其呈悬浮液。加入1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCl) 1.9 g和1-羟基苯并三唑(HOBt) 0.1 g,于室温反应0.5 h。加入(S) -6·HCl 1.8 g(10 mmol),反应24 h。加入二氯甲烷10 mL,用纯净水(2×15 mL)洗涤后减压蒸除溶剂得淡黄色固体1 3.9 g。用乙腈(约15 mL)溶解,通入氯化氢气体搅拌反应1 h。置冰箱中过夜,过滤,滤饼干燥得淡黄色晶体1 4.1 g,收率77.8%,m.p.223℃~226℃,[α]2D5+115°(c 10 mg·mL-1,甲醇) (226℃~228℃,+ 116.4°[5]) ;1H NMR δ: 1.48~2.72(m,6H),2.26(s,3H),2.28(s,3H),3.54 (s,3H),4.44 (s,2H),4.94 (s,1H),5.09(m,1H),7.3~8.02(m,9H),9.08 (s,1H) ; IR ν: 3 342,3 079,2 964,2 902,2 797,1 700,1 619,1 528,1 490,1 381,1 349,1 261,1 097,1 018,864,800,741,700,662 cm-1; ESI-MS: 492.2{[M + H]+}。
2 结果与讨论
(1)在Scheme 1路线中,合成关键中间体(R) -4时,可选择性水解,操作简便,反应完全,与路线一相比,双水解杂质大大减小。文献[3]方法合成(R) -4时,需要经过缩合、环合和水解3步反应。本文改进为一锅煮反应,减少操作;并改用四氢吡咯为催化剂,反应时间从19 h缩短至5 h,收率由56.3%提高至64.7%,无需复杂的柱层析纯化,纯度即达92.5%,可直接用于下一步反应。
(2)在合成(R) -4时,文献[9]方法使用辛可尼丁拆分剂。本文改用奎尼丁为拆分剂,并参考文献[10]方法调整溶剂比例,(R) -4直接析出,过滤后游离即可得到(R) -4。
(3)文献[5]方法在(S) -5与(S) -6·HCl酯化时,采用二氯亚砜为酰化剂进行缩合,反应副产物多,反应条件苛刻,过程不易控制。文献[3]方法对此进行改进,改为五氯化磷,但对环境污染大,操作不易。而本文采用EDCl和HOBt为缩合剂,于室温反应,具有反应操作简单,条件温和,且直接成盐合成1,收率从65.6%提高到77.8%,且无需柱色谱分离。此步酯化反应方法未见文献报道。
(4)将文献[4]方法合成(S) -5时,所用溶剂为甲苯。本文改用二甲苯为溶剂,产品纯度高,无需进一步重结晶,即可用于后续反应。
(5)将文献[11]方法在合成(S) -6时,后处理中增加酸洗,碱洗的过程,产品纯度由89%提高至95%以上。本文进一步将其制成盐酸盐得固体,利于长期保存,有利于工业化生产贮藏。
Scheme 1路线具有成本低、污染小、安全性高、收率高等优点。
参考文献
[1]P Buranakitjaroen,B Koanantaku,M Phoojaroenchanachai,et al.The efficacy and tolerability of barnidipine hydrochloride in Thai patients with hypertension [J].The Journal of International Medical Research,2004,32: 185-200.
[2]稻垣治,柴崎雅之,內田渡,等.新規カルシウム拮抗藥YM-09730-5の麻醉イヌの冠動脈及び心血管系に對する作用[J].基礎と臨床,1990,24:5621-5270.
[3]李立威,程志刚,李勋军,等.一种盐酸巴尼地平的合成[P].CN 101 643 469,2012.
[4]王丽,陈国华.盐酸巴尼地平的合成[J].化工时刊,2007,21(6) : 27-29.
[5]Kazuharu Tamazawa,Hideki Arima,Tadao Kojimal,et al.Stereoselectivity of a potent calcium anta gonist,1-benzyl-3-pyrrolidinyl methyl 2,6-dimethyl-4-(m-nitrophenyl ) -1,4-dihydro-pyridine-3,5-dicarboxylate [J].Journal of Medicinal Chemistry,1986,29(12) : 2504-2511.
[6]玉泽一治,有马英纪,冈田石,等.YM-09730非对映体A的右旋旋光异构体生产的一种新工艺[P].CN 85 107 590,1986.
[7]Zhi-gang Cheng,Xu-yong Dai,Li-wi Li,et al.Synthesis and characterization of impurities of Barnidipine hydrochloride,an antihypertensive drug substance[J].Molecules,2014,19: 1344-1352.
[8]刘益林,祁伟,杨琰.一种高纯盐酸乐卡地平的合成工艺[P].CN 102 516 160,2012.
[9]胡爱希,伍小云,黄红林.手性1,4-二氢-4-(3-硝基苯基) -3,5-吡啶二羧酸酯及其制备方法与应用[P].CN 101 357 901,2009.
[10]Bang-le Zhang,Wei Shi,Xin Shi,et al.Synthesis and biolo gical activity of the calciummodulator (R) and (S) -3-methyl-5-pentyl-2,6-dimethyl-4-(3-nitrophenyl) -1,4-dihydropyridine-3,5-dicarboxylate[J].Bioor ganic&Medicinal Chemistry Letters,2010,20: 805-808.
[11]Oldevilla,Madrid,Nurla.Process for preparing 3-substituted pyrrolidine compounds cross-reference to a related application[P].WO 153 649A1,2009.
·制药技术·
Process Improvement on the Synthesis of Barnidipine Hydrochloride
ZHOU Meng-yue1,JIANG Chuan-liang2,CHEN Guo-hua1
(1.Department of Medicinal Chemistry,China Pharmaceutical University,Nanjing 210009,China; 2.Nanjing Tianhai Pharmaceutical Technology Co.,Ltd,Nanjing 210048,China)
Abstract:A key intermediate,2,6-dimethyl-4-(m-nitrophenyl) -1,4-dihydropyridine-3-carboxylic acid-5-carboxylate(4),was prepared by reaction of 3-hydroxy-propionitril with diketene,then reacted with m-nitrobenzaldehyde and methyl 3-aminocrotonate.(R) -4 was obtained by resolution of 4 using quinidine as the resolution agent.Barnidipine hydrochloride in total yield of 12.7% was synthesized by esterification of (R) -4 with (S) -N-benzyl-3-hydroxy-pyrrolidine,then salifying.The structure was confirmed by1H NMR,IR and ESI-MS.
Keywords:Barnidipine hydrochloride; 1,4-dihydropyridine; drug synthesis; process improvement
作者简介:周梦月(1990-),女,汉族,江苏南京人,硕士研究生,主要从事药物合成的研究。E-mail: xiahua680712@163.com
*收稿日期:2015-01-10;
修订日期:2015-04-20
中图分类号:R914.5; O621.3
文献标识码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.07.0653
通信联系人:陈国华,副研究员,Tel.025-83241246,E-mail: cgh63@163.com
猜你喜欢
四川大学学报(自然科学版)(2025年1期)2025-02-10 00:00:00
——非均布滤饼的局部比阻与平均比阻的测定与计算方法
化工装备技术(2020年4期)2020-09-09 07:34:10
流体机械(2020年5期)2020-06-24 05:39:08
石油钻探技术(2018年5期)2018-10-13 07:25:18
中国畜牧兽医文摘(2018年6期)2018-07-13 10:21:02
中国矿业(2017年2期)2017-02-28 02:12:56
广东饲料(2016年6期)2016-12-01 03:43:29
畜牧兽医科技信息(2016年4期)2016-11-29 09:18:31
今日畜牧兽医(2015年3期)2015-04-17 00:58:40
应用化工(2014年4期)2014-08-16 13:23:09
