
双苯噁唑酸的合成工艺改进*
2015-02-02 07:53:37胡乐乐陈志卫浙江工业大学药学院绿色制药技术与装备教育部重点实验室浙江杭州310014
合成化学 2015年7期
井 蕾,胡乐乐,陈志卫(浙江工业大学药学院绿色制药技术与装备教育部重点实验室,浙江杭州 310014)
双苯噁唑酸的合成工艺改进*
井蕾,胡乐乐,陈志卫
(浙江工业大学药学院绿色制药技术与装备教育部重点实验室,浙江杭州310014)
摘要:以甘氨酸为原料,经酯化和肟化反应制得2-氯-2-肟乙酸乙酯(3) ;以1,3-二甲基-1,3-二(磺酸丁基)咪唑啉酮硫酸氢根功能性离子液体{[DBSDMI]2HSO4}为催化剂,3与1,1-二苯基乙烯经1,3-偶极环加成反应合成了双苯噁唑酸,总收率63.2%,其结构经1H NMR,13C NMR和ESI-MS确证。
关键词:双苯噁唑酸; 1,1-二苯基乙烯;合成;工艺改进
双苯噁唑酸(1)是由安万特公司研究开发的异唑类安全剂,主要用于防除玉米田间一年及常年生杂草等。1的使用不仅提高作物的耐药性,也解决难除杂草的防除问题,扩大了除草剂的应用范围。
目前1的合成路线主要有两条:路线一:以乙醚为溶剂,1,1-二苯基乙烯(4)和2-氯-2-肟乙酸乙酯(3)在三乙胺催化下制备[2]。该路线的主要缺点是使用低沸点溶剂乙醚,易燃易爆,存在安全隐患。路线二:以乙醇为溶剂,4和2-硝基乙酸乙酯在1,4-二氮杂二环[2.2.2]辛烷(DABCO)催化下制备[3]。该路线的主要缺点是硝基乙酸乙酯不容易制备而且价格昂贵。
本文以路线一为基础合成1(Scheme 1),但对其工艺条件进行改进: (1)以甘氨酸为原料,经酯化和肟化反应制得3,收率从文献[4]方法的54%提高至81%; (2)在4的合成中,以溴苯和镁屑在2-甲基四氢呋喃中先制备成苯基溴化镁格氏试剂,然后与苯乙酮经一锅法制得4,收率高达95.6%; (3)在1的合成中,采用2-甲基四氢呋喃替代文献方法中的环己烷[5]或者乙醚[2],收率从64%[5]提高至82.5%。
改进后的工艺条件具有反应条件温和、操作简便,总收率较高(63.2%)等优点,有利于工业化生产。
1 实验部分
1.1仪器与试剂
WRS-1A型数字熔点仪(温度未校正) ; Varian-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标) ; Trace DSQ FINNIGSN型质谱仪。
1,3-二甲基-1,3-二(磺酸丁基)咪唑啉酮硫酸氢根功能性离子液体{[DBSDMI]2HSO4}按文献[6]方法合成;其余所用试剂均为分析纯。
1.2合成
(1) 4的合成
在反应瓶中加入镁屑9.95 g(0.41 mol),1,2-二溴乙烷1.47 g(8 mmol)及无水2-甲基四氢呋喃65 mL,通氮气,密封装置,搅拌下升温至65℃引发反应,缓慢滴加溴苯61.23 g(0.39 mol),滴毕,于65℃反应4 h制得苯基溴化镁格氏试剂。控制内温≤20℃,缓慢滴加苯乙酮44.6 g(0.37 mol)的2-甲基四氢呋喃(30 mL)溶液,滴毕,于80℃反应3 h。冷却至5℃,用浓盐酸调至pH 6~7,加入催化剂[DBSDMI]2HSO41.08 g,回流脱水反应2 h。常压回收2-甲基四氢呋喃,残留物高真空蒸馏,收集(106~110)℃/267 Pa馏分得无色油状液体4 62.2 g,收率95.6%,b.p.277℃/ 101.3 kPa (277.1℃[8]),含量99.1% (GC) ;1H NMR δ: 7.31(s,10H,PhH),5.4(s,2H,CH2) ;13C NMR δ: 150.1(C2),141.5,128.2,127.7 (ArC),114.2 (CH2) ; ESI-MS m/z: 157{[M-Na]-}。
(2)氨基乙酸乙酯盐酸盐(2)的合成
在四口瓶中加入甘氨酸75 g(1 mol)和乙醇400 mL,搅拌使其溶解;于-10℃缓慢滴加氯化亚砜142.8 g(1.2 mol),滴毕,回流(80℃)反应4 h。冷却至室温,过滤,滤饼真空干燥得白色固体2 129.7 g,收率98.9%,m.p.144℃~146℃(145℃~148℃[7]) ;1H NMR(DMSO-d6)δ: 8.50(s,3H,NH3),4.17(q,J = 7.2 Hz,2H,OCH2),3.74(s,2H,COCH2),1.22(t,J =7.4 Hz,3H,CH3) ; ESI-MS m/z: 126{[M + Na+}。
(3) 3的合成
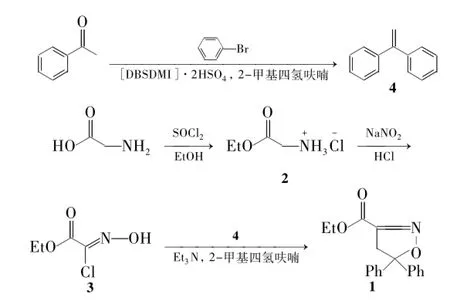
Scheme 1
在四口烧瓶中加入2 31 g(0.22 mol)。搅拌下加入浓盐酸15.1 mL和水47 mL,于-10℃滴加亚硝酸钠12.3 g的水(20 mL)溶液,滴毕(约30 min),反应30 min(操作A)。重复操作A两次,于0℃~5℃反应1 h。用二氯甲烷(3×40 mL)萃取,合并萃取液,用无水硫酸钠干燥,常压回收二氯甲烷,残余物用环己烷重结晶得白色固体3 26.9 g,收率81%,m.p.79℃~81℃(80℃[4]) ;1H NMR δ: 9.56(s,1H,OH),4.35(q,J =7.2 Hz,2H,CH2),1.38(t,J =7.2 Hz,3H,CH3)。
(4) 1的合成
在三口烧瓶中加入4 36.7 g(0.2 mol),三乙胺18.9 g(0.19 mol)及2-甲基四氢呋喃50 mL,搅拌下滴加3 25.8 g的2-甲基四氢呋喃(60 mL)溶液,滴毕(2.5 h~3.0 h),于室温反应3 h。减压回收溶剂,加水100 mL,二氯甲烷(3×50 mL)萃取,合并萃取液,减压蒸馏回收二氯甲烷,残余物经硅胶柱层析[洗脱剂: V(石油醚)∶V(乙酸乙酯) = 16∶1]纯化得白色固体1 41.6 g,收率82.5%,纯度99.1%,m.p.86.5℃~88.0℃(87℃~88℃[3]) ;1H NMR δ: 7.30~7.50(m,10H,PhH),4.33(q,J = 7.2 Hz,2H,OCH2),3.85 (s,2H,CH2),1.36(t,J =7.2 Hz,3H,CH3) ;13C NMR δ: 160.4,151.0,142.9,128.5,127.9,125.6,94.6,61.9,46.6,13.9; ESI-MS m/z: 296{[M +H]+}。
2 结果与讨论
2.1合成
(1) 3的合成
在3的合成中,文献[4]方法采用-5℃下,两批次滴加HCl/NaNO2溶液的方法。本文改进为-10℃下,分三批次滴加HCl/NaNO2溶液。考虑到反应过程中会有部分HNO2不稳定而分解,还将HCl/NaNO2的用量由文献[4]方法的2.0 eq.提高至2.4 eq.,收率从54%[4]提高至81%。
(2) 4的合成
在4的合成中,本文用2-甲基四氢呋喃替代文献方法中的乙醚[9-11],四氢呋喃[12]作为溶剂制备格氏试剂,反应过程中可明显减少副产物联苯的生成。苯基溴化镁的收率由82%[11]提高至99%。
文献[9-10,13-14]方法合成4时均采用分步法,即首先制得1,1-二苯基乙醇,然后回流脱水得4。考虑到文献[13-14]方法中甲基氯化镁成本较高,本文对文献[9-10]方法进行优化改进。在实验中发现,在苯基溴化镁与苯乙酮的亲核加成过程中,已经有部分4生成。水解得到1,1-二苯基乙醇过程也会有较多的1,1-二苯基乙醇脱水生成4。文献[9]方法先通过重结晶得到1,1-二苯基乙醇,然后再脱水制备4时必然会造成产物大量损失。笔者考虑到2-甲基四氢呋喃具有沸点适中、与水不互溶、分层明显等优点同时适合做制备格式试剂和脱水反应的溶剂,用它替代文献中乙醚、四氢呋喃和甲苯,实现了中间体1,1-二苯基乙醇不经分离纯化直接一锅法合成4。本工艺不仅解决了分步法中产物损失的问题,而且与传统工艺[9-10,13-14]相比,该工艺还具有路线步骤简短、条件温和、操作简便、成本低等优点,总收率更高达95.6%。
(3) 1的合成
在1的合成中,本文使用2-甲基四氢呋喃替代文献方法中的环己烷[5]、乙醚[2],不仅降低了工业生成过程中的危险,而且收率由64%[5]提高至82.5%。而且2-甲基四氢呋喃为溶剂后处理简便、回收低能耗使得该工艺更加适合工业化生产。
参考文献
[1]邓金保.拜耳与美国杜邦公司之间开展的除草剂交易[J].农化新世纪,2007,(4) : 35.
[2]L威尔姆斯,K鲍尔,H比林格.取代异恶唑啉及其制备方法,含有它们的试剂含其作为防护剂的用途[P].CN 1 133 038,1996.
[3]Cremonesi G,Croce P D,Fontana F,et al.Stereoselective synthesis of β,ε-dihydroxy-α-amino acids by ring opening of 4,5-dihydroisoxazolyl derivatives[J].Tetrahedron,2008,19(24) : 2850-2855.
[4]Kozikowski A P,Adamczyk M.Methods for the stereoselective cis-cyanohydroxylation and carboxyhydroxylation of olefins[J].J Org Chem,1983,48(3) :366-372.
[5]王慧,周月跟,王广宇,等.安全剂双苯噁唑酸的合成[J].农药,2012,51(11) : 792-793.
[6]Chen Z W,Zhu Q,Su W K.A novel sulfonic acid functionalized ionic liquid catalyzed multicomponent synthesis of 10,11-dihydrochromeno[4,3-b]chromene-6,8-(7H,9H) -dione derivatives in water[J].Tetrahedron Lett,2011,52(20) : 2601-2604.
[7]Chambers R W,Carpenter F H.On the preparation and froperties of some amino acid amides[J].J Am Chem Soc,1955,77(6) : 1522-1526.
[8]Serigan K T,Wise P H.Dicyclic hydrocarbons.III.Diphenyl-and dicyclohexylalkanes through C15[J].J Am Chem Soc,1951,73(10) : 4766-4769.
[9]李祥高,吴安树,何莉莉,等.1,1-二芳基-3-卤代丙烯的制备方法[P].CN 1 651 371,2004.
[10]王江,张卫.格氏反应制备1,1-二苯乙烯[J].河北化工,2000,(4) : 14-15.
[11]Martin L J,Spivey A C,Ellames G J,et al.The devolatilization of[14C]-labelled aromatics towards the synthesis of[14C]-labelled NK antagonists for PK studies[J].Journal of Labelled Compounds and Radiopharmaceuticals,2007,50(5-6) : 607-608.
[12]Zhu M Y,Liu J Q,Yu J J,et al.AlCl3-promoted formal[2 +3]-cyclo addition of 1,1-cyclopropane diesters with N-benzylic sulfonamides to construct highly stereoselective indane derivatives[J].Org Lett,2014,16(17) : 1856-1859.
[13]Suzuki,Ken.2,2-(Diaryl) vinyl-phospine compond,palladium catalyst thereof,and process for producing arylamine,diaryl,or arylalkyne with the catalyst[P].EP 1 167 372,2001.
[14]李年康.医药中间体二苯基乙烯的合成[J].化学工程与装备,2012,(11) : 20-21.
·研究简报·
Process Improvement on the Synthesis of Isoxadifen-ethyl
JING Lei,HU Le-le,CHEN Zhi-wei
(Key Laboratory for Green Pharmaceutical Technologies and Related Equipment of Ministry of Education,College of Pharmacy,Zhejiang University of Technology,Hangzhou 310014,China)
Abstract:Isoxadifen-ethyl in total yield of 63.2% was synthesized by 1,3-dipolar cycloaddition of 1,1-diphenyl ethylene with 2-chloro-2-oxime ethyl acetate,which was obtained by esterification and oximation,using glycine as the material and[DBSDMI]2HSO4as the catalyst.The structure was confirmed by1H NMR,13C NMR and ESI-MS.
Keywords:Isoxadifen-ethyl; 1,1-diphenyl ethylene; synthesis; process improvement
作者简介:井蕾(1988-),男,汉族,河南信阳人,硕士研究生,主要从事医药中间体的合成研究。
基金项目:浙江省科技厅资助项目(2012R10043-08 )
*收稿日期:2014-10-08;
修订日期:2015-05-10
中图分类号:O626.2
文献标识码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2015.07.0630
通信联系人:陈志卫,副研究员,硕士生导师,Tel.0571-88871087,E-mail: chenzhiwei@ zjut.edu.cn
猜你喜欢
化工设计(2022年4期)2023-01-02 17:44:05
昆明医科大学学报(2021年8期)2021-08-13 08:59:14
山东化工(2019年7期)2019-04-27 07:39:28
中成药(2018年6期)2018-07-11 03:01:04
中国资源综合利用(2017年1期)2018-01-22 02:44:25
化工管理(2017年35期)2018-01-10 11:19:56
中成药(2017年8期)2017-11-22 03:19:25
中国塑料(2017年2期)2017-05-17 06:13:27
天然产物研究与开发(2016年11期)2016-06-15 20:29:17
苏州科技大学学报(工程技术版)(2015年3期)2015-02-28 16:21:12
