
石墨烯/纳米管复合结构吸附分离CO2/CH4混合物的分子模拟
2015-01-04 12:52:08雷广平重庆大学低品位能源利用技术及系统教育部重点实验室重庆400030
物理化学学报 2015年4期
雷广平 刘 朝 解 辉(重庆大学低品位能源利用技术及系统教育部重点实验室,重庆400030)
石墨烯/纳米管复合结构吸附分离CO2/CH4混合物的分子模拟
雷广平 刘 朝*解 辉
(重庆大学低品位能源利用技术及系统教育部重点实验室,重庆400030)
采用巨正则蒙特卡洛(GCMC)及分子动力学(MD)方法探讨了石墨烯/碳纳米管三维骨架结构(GNHS)对等摩尔CO2/CH4二元混合物的吸附分离性能.模拟结果表明CO2比CH4更易吸附于GNHS中,GNHS与(6,6) SWCNT(单壁碳纳米管)相比具有更高的分离性能.随着温度升高,CO2的吸附量快速降低,而CH4的吸附量则呈现出先升高后降低的趋势.最后采用分子动力学方法计算了CO2与CH4的自扩散系数及停留时间等动力学相关参数,发现CO2在GNHS内扩散的阻力更大.而各组分在吸附剂外部吸附层内的扩散过程对混合物的分离也存在一定影响.
石墨烯/碳纳米管三维骨架结构;吸附等温线;停留时间;吸附选择性;吸附系数
1 引言
天然气作为一种经济、清洁的化石能源,被广泛地应用于各个领域.通常天然气含有少量CO2, H2S,N2等杂质.1如我国川东北地区开采的天然气中含有高达15%(体积分数)的H2S气体,CO2含量也接近10%.2杂质的存在不仅降低了天然气的热值, CO2、H2S等酸性气体还会造成设备及输送管道的腐蚀,因此在输送和使用前必须将其除去.传统脱硫脱碳方法主要是物理及化学吸收法,2,3这些方法能耗高、污染大,吸收剂再生困难.分子筛、4-6金属有机骨架7,8及无机碳材料9-13等多孔材料用于吸附分离气体混合物则具有绿色环保、能耗低、再生容易等优点而得到越来越多的关注.
©Editorial office ofActa Physico-Chimica Sinica
碳纳米管及石墨烯等无机碳材料具有高比表面积、耐腐蚀、高化学稳定性及热稳定性等优点,可作为一种理想的吸附材料.14-17近年来,科学家成功合成出石墨烯/碳纳米管三维骨架结构(GNHS)18-20并发现其具有良好的导热、机械、21导电22及吸附性能.23,24研究发现GNHS具有很高的储氢能力,当掺杂有锂离子时GNHS的储氢能力可达到41 g·L-1.23GNHS还是一种优良的气体分离膜,可有效分离惰性气体混合物.25然而,GNHS作为一种吸附剂用于分离气体混合物还未见报道.
本文采用巨正则蒙特卡洛(GCMC)方法,探讨压力、温度以及气相组成对CO2/CH4二元混合物在GNHS中吸附分离性能的影响.并采用分子动力学(MD)方法研究了CO2及CH4在GNHS中的扩散性能、停留时间及吸附系数等动力学性质.
2 模拟细节
本文中GNHS采用Xu等21所提出的模型. GNHS由相互垂直的(6,6)CNT和多孔石墨烯搭建而成,如图1(a)所示.首先,将(6,6)CNT垂直放置于去除了24个碳原子(图1(b)中圆内的原子)的石墨烯平板上.然后采用Materials Studio(MS)中的DMol3模块对所得结构进行几何优化.优化后的结构中,石墨烯与CNT连接处形成了六个七元环(图1(c)中环A).最后,对所得结构沿三个方向扩展即得到GNHS.
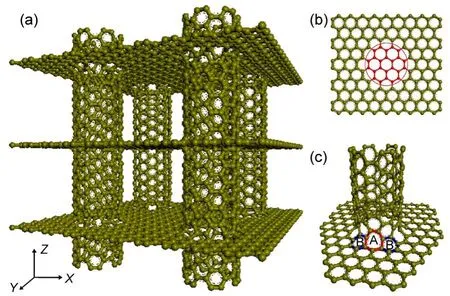
图1 GNHS示意图Fig.1 Schematic of the GNHS
分别采用GCMC及MD方法研究了CO2/CH4混合物在GNHS中的吸附分离性能及动力学性质.模拟中CH4与CO2均采用全原子模型,短程范德华力采用Lennard-Jones(L-J)模型,长程作用采用Ewald法进行计算,截断半径选取为1.2 nm.吸附剂尺寸为4.064 nm×4.401 nm×2.656 nm,碳纳米管在X、Y方向的距离分别为2.032和2.198 nm,石墨烯狭缝宽度保持为0.664 nm.模拟中吸附剂材料保持刚性结构, X、Y、Z三个方向均采用周期性边界条件.各势能模型参数如表1所示.26
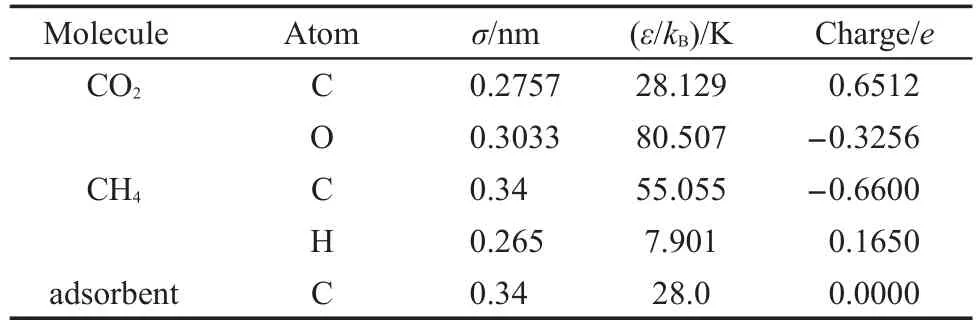
表1 各势能模型的Lennard-Jones(L-J)参数及电荷26Table 1 Lennard-Jones(L-J)parameters andcharges for all potential models26
GCMC模拟采用Music软件27进行,模拟中按照等权重方法对分子进行平动、转动、插入和删除四种移动.对各种尝试移动接受概率的详细介绍见文献.28Peng-Robinson(PR)状态方程用于计算气相主体中各组分的逸度,交互作用因子为0.0919.1每次计算共进行1×108步,其中前5×107步用于体系平衡计算,后5×107步用于统计相关数据.
通过GCMC模拟可得到各组分的吸附量和吸附选择性,其中计算吸附选择性的方程如下:17

式中,x、y分别表示气相主体及吸附剂中CO2和CH4的摩尔分数.
最后采用MD方法研究了温度300 K和压力3.0 MPa时,CO2与CH4在GNHS中的吸附动力学性质.为了使系统保持恒温恒压,首先采用GCMC模拟得到给定温压下的最小能量构型,并采用该构型作为MD模拟的初始结构.模拟快照如图2所示,图2(a)结构用于计算CO2与CH4两组分在GNHS中的扩散系数及停留时间,图2(b)结构用于计算两组分的吸附系数及吸附时间.两模拟系统尺寸分别为4.064 nm×4.401 nm×2.656 nm和8.0 nm×8.0 nm×8.0nm,模拟均采用lammps软件进行,系综选取NVT系综,并采用Nose-Hoover方法使温度维持在300 K.时间步长设为2 fs.所有模拟计算时间均为3.0 ns,其中前1.0 ns用于系统平衡计算,后2.0 ns用于数据的统计.

图2 停留时间和自扩散系数计算模型(a)和各组分的吸附时间及吸附系数计算模型(b)的模拟快照Fig.2 Snapshots of simulations for calculation of residence time and self-diffusivity(a)and calculation of adsorption time and adsorption coefficient for every component(b)
3 结果与讨论
3.1 压力对等摩尔CO2/CH4混合物吸附分离性能的影响
为了验证CH4全原子模型的准确性,我们首先与文献26中CH4在ACF-15吸附剂中的吸附结果进行了比较,如图3所示.我们的模拟结果与文献26符合很好.
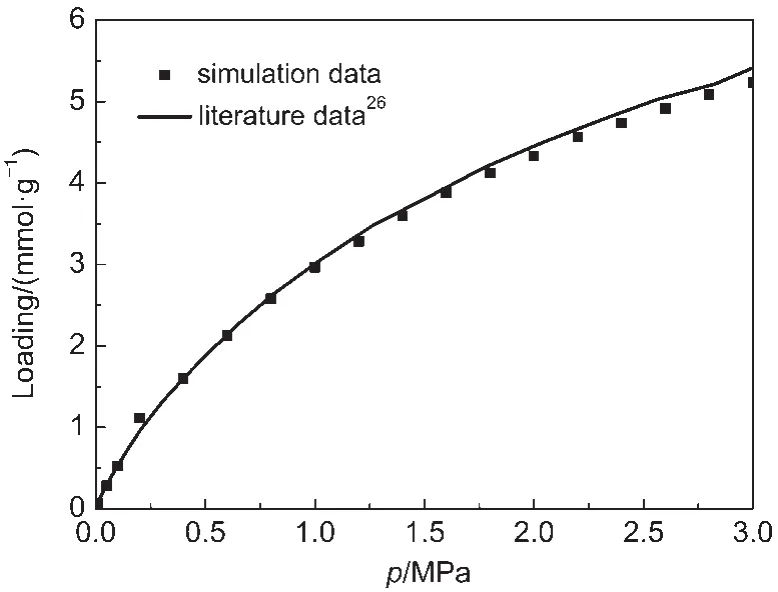
图3 300 K时CH4在ACF-15中的吸附等温线Fig.3 Adsorption isotherms of CH4inACF-15 at 300 K
首先对等摩尔CO2/CH4混合物在GNHS及(6,6) SWCNT中的吸附分离性能进行了比较研究.图4给出了CO2与CH4在两种不同吸附剂内的吸附等温线.两组分在吸附过程中存在竞争且两种吸附剂对CO2均具有较高的吸附能力,但SWCNT对CO2的吸附能力远低于GNHS.对CH4,情况则相反,SWCNT对CH4的吸附能力高于GNHS.可以从图5来探讨出现这一现象的原因,图5(a)表明两吸附质组分与(6, 6)SWCNT间的相互作用能明显高于其与GNHS间的相互作用能,这是因为弯曲碳纳米管对其内部吸附质分子作用能的叠加所致,与Liu等26的研究结果一致.但是吸附质与吸附剂间的相互作用能无法解释两吸附质组分在GNHS及(6,6)SWCNT内部吸附量的变化.图5(b)给出了吸附质与吸附质间的相互作用与压力变化的关系,可以发现吸附质与吸附质间的相互作用能随着压力升高而增强,与吸附量的变化规律一致.SWCNT内CH4与吸附质间相互作用能高于GNHS内部CH4与吸附质间相互作用能,而CO2与吸附质间相互作用能则相反.这表明随着压力变化,气体组分的吸附量主要受到吸附质分子间相互作用的影响.
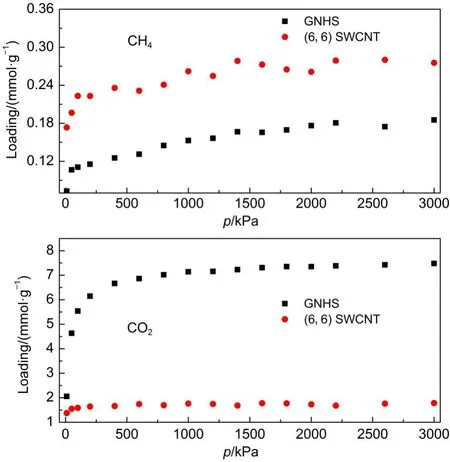
图4 等摩尔CO2/CH4混合物中各组分在GNHS和(6,6)SWCNT中的吸附等温线Fig.4 Adsorption isotherms of each component in equimolar CH4/CO2mixture in the GNHS and(6,6)SWCNT
为了进一步探讨SWCNT与GNHS吸附剂对气体混合物的分离能力,图6给出了CO2的选择性与压力的关系,可知GNHS比(6,6)SWCNT具有更高的分离性能.GNHS对CO2的选择性随着压力升高出现先升高后降低至一稳定值的趋势.这主要是随着压力升高,CO2吸附量首先达到饱和状态,而CH4的吸附量则还在继续升高所致(如图4所示).
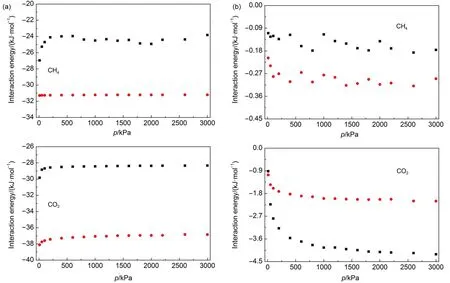
图5 吸附质与吸附剂之间(a)及吸附质间(b)的相互作用能与压力关系Fig.5 Relation between interaction energy of adsorbate-adsorbent(a),adsorbate-adsorbate(b)and pressure

图6 GNHS和(6,6)SWCNT中CO2的选择性与压力关系Fig.6 Relation between selectivity of CO2in the GNHS and(6,6)SWCNT and pressure
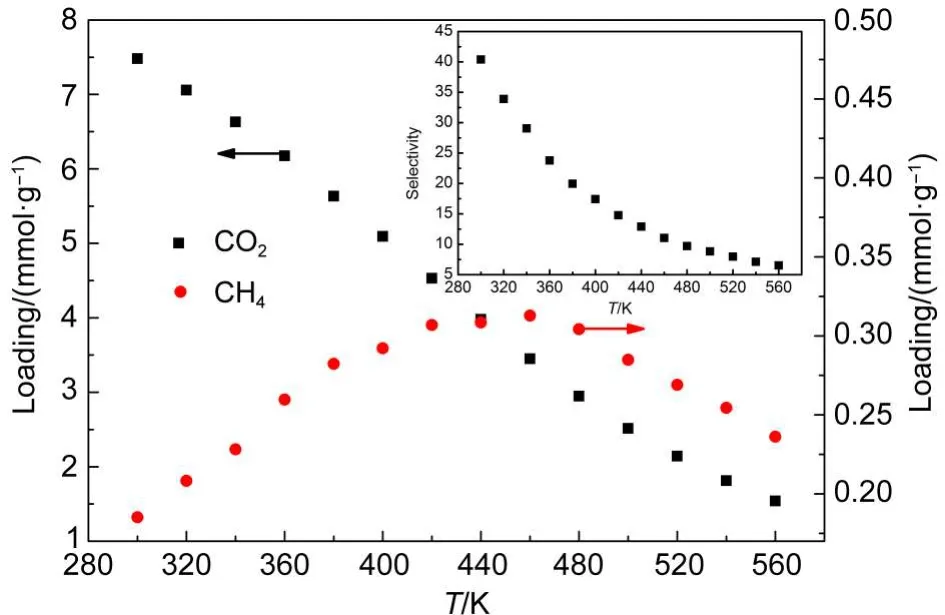
图7 CO2/CH4混合物中各组分的吸附量及CO2选择性与温度关系Fig.7 Relation of loading for each component in CO2/CH4mixture and selectivity of CO2with temperature
3.2 温度及气相组成对等摩尔CO2/CH4混合物吸附分离性能的影响
由于GNHS相对于(6,6)SWCNT具有更好的分离性能,而且本文主要目的是探讨各影响因素对GNHS吸附分离性能的影响,因此下面仅对GNHS吸附分离性能进行讨论分析.图7给出了压力3.0 MPa时,两组分在GNHS内部吸附量随温度的变化关系.在所研究的温度范围内,CO2吸附量均高于CH4.随着温度升高,CO2吸附量持续降低,而CH4吸附量则呈现出先升高后降低的趋势.这同样可以通过图8所示的吸附质间相互作用能来解释,吸附质间相互作用能随温度的变化趋势与吸附量的变化趋势是一致的.可能是随着温度升高,吸附质分子的热运动增强,吸附质分子间距离增大,两分子间相互作用能降低,从而使得更多CO2分子发生脱附过程,而CO2的脱附又使得吸附剂内部空位数增多,从而增加了CH4进入吸附剂内部的可能性.随着温度升高,CO2选择性持续降低,对混合物的分离有着不利影响.
图9给出了气相中CO2含量对吸附量及选择性的影响.随着气相中CO2含量降低,CH4吸附量逐渐升高.但是即使CO2在气相中含量很低时,CO2吸附量也高于CH4,当CO2含量为0.1时,其选择性可达到70左右.说明GNHS可有效分离含低浓度CO2的天然气混合物.
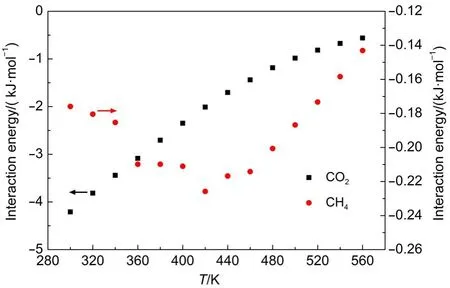
图8 吸附质间相互作用能与温度关系Fig.8 Relation between interaction energy of adsorbate-adsorbate and temperature
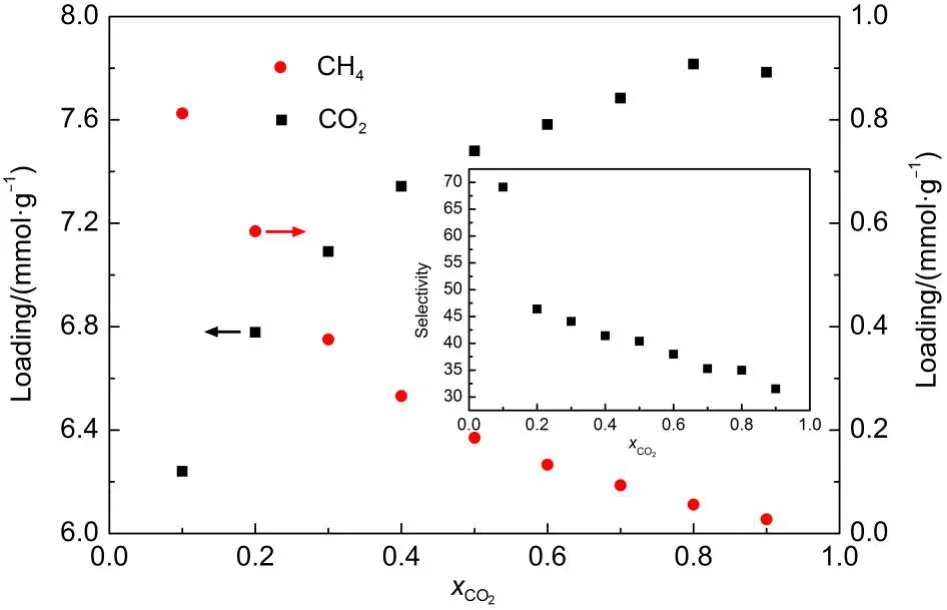
图9 CO2/CH4混合物中两组分吸附量及CO2选择性随CO2组成的变化关系Fig.9 Variation of loading of two components in CO2/CH4mixture and selectivity of CO2with fraction of CO2
3.3 CO2/CH4混合物在GNHS内的吸附动力学性质
采用分子动力学方法探讨了两组分在GNHS中的动力学性质.两组分在GNHS内的均方位移(MSD)如图10所示.由于在体系中CH4含量比较低,得到的MSD曲线起伏较大,但两组分的MSD基本符合线性变化.根据方程(2)计算得到CO2与CH4的自扩散系数分别为8.994×10-10和2.617×10-9m2·s-1, CH4在GNHS内扩散能力明显高于CO2.

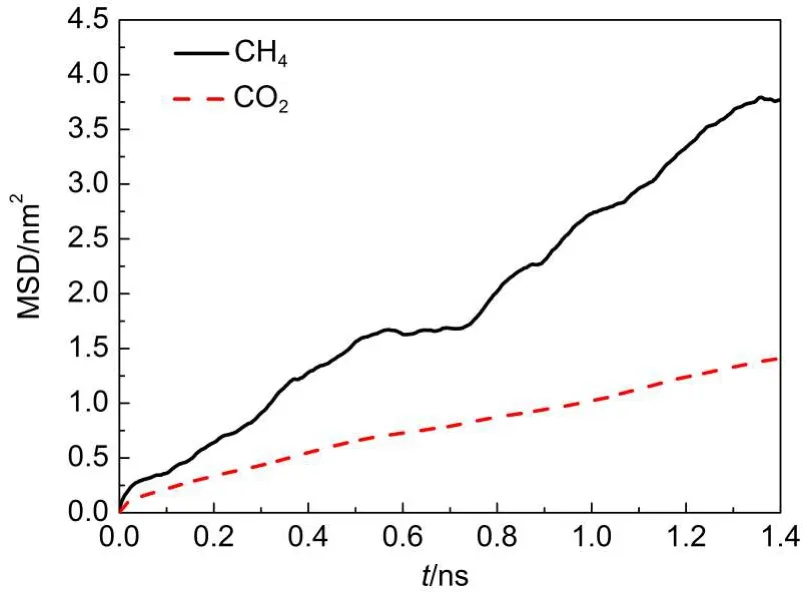
图10 CO2与CH4在GNHS内的均方位移(MSD)Fig.10 Mean-square displacements(MSD)of CO2and CH4in the GNHS
上式中D为自扩散系数,t表示时间,r(0)表示初始时刻位置,r(t)表示t时刻位置.
为了进一步探讨吸附质分子与吸附剂之间的相互作用,图11给出了两组分与GNHS的径向分布函数.CH4与CO2径向分布函数的第一吸附层位置分别为0.556和0.675 nm.我们以CO2与CH4第一吸附层位置作为判断条件(rc)计算两组分分子在GNHS材料中某特定原子处的吸附停留时间自相关函数.原子i在原子j处的停留时间自相关函数(Cres(t))如方程(3)所示:29

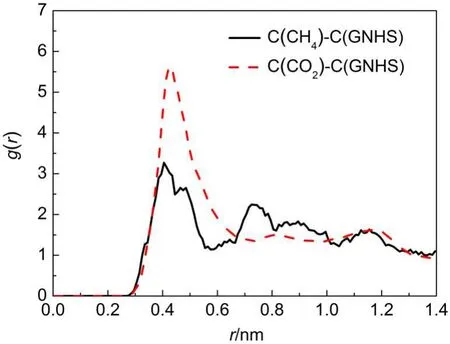
图11 CO2-GNHS及CH4-GNHS的径向分布函数Fig.11 Radial distribution functions of CO2-GNHS and CH4-GNHS
式中,当原子i与j之间距离在0与t时刻均小于判断条件rc时,nij(t)=1;否则nij(t)=0.根据定义可知存在两种不同的停留时间自相关函数,一种称为连续停留时间自相关函数即在时间间隔[0,t]内原子i与j之间距离始终小于rc;另一种称为间歇停留时间自相关函数在这种情况中,原子i与j之间距离只需在0与t时刻小于rc而不考虑时间间隔(0,t)内的情况.对停留时间自相关函数积分后便得到分子的停留时间τres,如方程(4)所示.因此,对应的停留时间是连续停留时间和间歇停留时间

图12给出了CO2与CH4分子的停留时间自相关函数曲线.相对于CH4,CO2的连续及间歇停留时间自相关函数曲线均下降缓慢,说明CO2分子与GNHS材料具有更强的相互作用,CO2分子在GNHS中某一特定原子附近停留的时间较长,相应的连续与间歇停留时间分别为0.593和5.305 ns.而CH4分子的连续和间歇停留时间则分别为0.125和1.893 ns.因此,可以采用技术手段(如引入某种官能团)来增大各组分在多孔材料内停留时间的差异,使气体混合物的分离能力得以提高,关于这一方面的研究我们将在后续工作中进行.
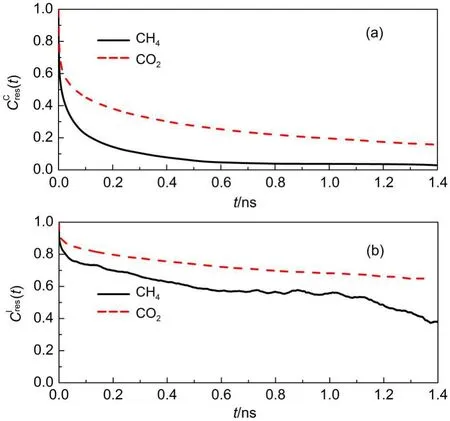
图12 CO2与CH4分子的连续停留时间自相关函数(CCres(t)) (a)及间歇停留时间自相关函数(CrIes(t))(b)Fig.12 Continuous(CCres(t))(a)and intermittent(CrIes(t))(b) residence time correlation functions of CO2and CH4
为了计算两组分的吸附系数及吸附时间,统计了两组分沿X方向的数密度分布,如图13所示.在GNHS外存在一吸附层(实线与虚线之间),气相主体中分子只有越过该吸附层才可进入GNHS内部.吸附系数α定义为从气相主体进入到GNHS内部的分子数NGNHS与进入到吸附层内的分子数NLayer之比,30如方程(5)所示.

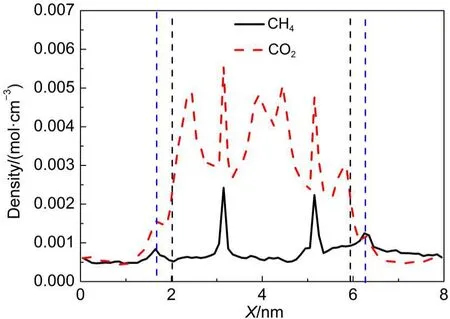
图13 CO2与CH4沿X方向的数密度分布Fig.13 Number density profile of CO2and CH4in X direction
吸附时间则为分子穿过吸附层所需要的时间.计算结果表明,300 K和3 MPa时,CH4的吸附系数与吸附时间分别为0.0131和21.76 ps,均低于CO2的吸附系数(0.0195)和吸附时间(45.48 ps),说明CH4分子可以更快速地从气相主体进入到吸附剂材料中.当CO2/CH4混合物与固体材料接触时间较短时,在吸附剂内部则会吸附较多的CH4分子,而当接触时间较长时,由于CO2与GNHS之间更强的相互作用,进入吸附层的CO2分子则更容易进入到GNHS内部从而取代已吸附的CH4分子.该现象表明吸附剂外侧的吸附层对气体混合物的吸附分离也具有一定的影响.
4 结论
本文采用GCMC与MD方法探讨了CO2/CH4二元混合物在GNHS多孔材料中的吸附分离性能.结果表明GNHS多孔材料比(6,6)SWCNT具有更好的分离性能.CO2在GNHS内的吸附量随着压力降低和温度升高而迅速降低,说明GNHS多孔材料易于再生.此外,采用MD方法研究发现CO2在GNHS内的自扩散系数为(8.994×10-10m2·s-1)低于CH4的自扩散系数(2.617×10-9m2·s-1),而其连续与间歇停留时间则均高于CH4,说明CO2在GNHS内部的扩散受到更大的阻碍作用.因此,可以通过增大气体混合物各组分在多孔材料内停留时间的差异来提高分离能力.对两组分吸附系数和吸附时间进行了计算,结果表明CH4的吸附时间(21.76 ps)低于CO2的吸附时间(45.48 ps),说明CH4可以更容易地穿过吸附层进入到GNHS吸附剂内部.由于CH4分子与GNHS间的相互作用较弱,因此CH4的吸附系数(0.0131)低于CO2的吸附系数(0.0195).
(1) Peng,X.;Cao,D.P.AIChE Journal 2013,59,2928.doi: 10.1002/aic.v59.8
(2) Zhang,J.H.;Shen,B.X.;Sun,H.;Liu,J.C.Petroleum Science and Technology 2011,29,48.doi:10.1080/10916460903330221
(3) Krischan,J.;Makaruk,A.;Harasek,M.Journal of Hazardous Materials 2012,215-216,49.
(4) Ma,Y.H.;Zhao,H.L.;Tang,S.J.;Hu,J.;Liu,H.L.Acta Phys.-Chim.Sin.2011,27,689.[马燕辉,赵会玲,唐圣杰,胡 军,刘洪来.物理化学学报,2011,27,689.]doi:10.3866/ PKU.WHXB20110335
(5) Cosoli,P.;Ferrone,M.;Pricl,S.;Fermeglia,M.Chemical Engineering Journal 2008,145,93.doi:10.1016/j. cej.2008.08.013
(6) Chen,S.J.;Dai,W.;Luo,J.S.;Tang,Y.J.;Wang,C.Y.;Sun,W. G.Acta Phys.-Chim.Sin.2009,25,285.[陈善俊,戴 伟,罗江山,唐永建,王朝阳,孙卫国.物理化学学报,2009,25,285.] doi:10.3866/PKU.WHXB2009021
(7) Wu,X.J.;Zheng,J.;Li,J.;Cai,W.Q.Acta Phys.-Chim.Sin. 2013,29,2207.[吴选军,郑 佶,李 江,蔡卫权.物理化学学报,2013,29,2207.]doi:10.3866/PKU.WHXB201307191
(8) An,X.H,;Liu,D.H.;Zhong,C.L.Acta Phys.-Chim.Sin.2011, 27,553.[安晓辉,刘大欢,仲崇立.物理化学学报,2011,27, 553.]doi:10.3866/PKU.WHXB20110319
(9) Feng,W.;Kwon,S.;Borguet,E.;Vidic,R.Environmental Science&Technology 2005,39,9744.doi:10.1021/es0507158
(10) Arab,M.;Picaud,F.;Devel,M.;Ramseyer,C.;Girardet,C. Physical Review B 2004,69,165401.doi:10.1103/ PhysRevB.69.165401
(11) Huang,L.;Zhang,L.;Shao,Q.;Lu,L.;Lu,X.;Jiang,S.;Shen, W.The Journal of Physical Chemistry C 2007,111,11912.doi: 10.1021/jp067226u
(12) Jiang,J.;Sandler,S.I.Physical Review B 2003,68,245412. doi:10.1103/PhysRevB.68.245412
(13) Liu,L.;Bhatia,S.K.The Journal of Physical Chemistry C 2013,117,13479.doi:10.1021/jp403477y
(14) Razavi,S.S.;Hashemianzadeh,S.M.;Karimi,H.J.Mol. Model.2011,17,1163.doi:10.1007/s00894-010-0810-9
(15) Liu,X.Q.;Tian,Z.Y.;Chu,W.;Xue,Y.Acta Phys.-Chim.Sin. 2014,30,251.[刘晓强,田之悦,储 伟,薛 英.物理化学学报,2014,30,251.]doi:10.3866/PKU.WHXB201312243
(16) Ye,Q.;Zhang,Y.;Li,M.;Shi,Y.Acta Phys.-Chim.Sin.2012, 28,1223.[叶 青,张 瑜,李 茗,施 耀.物理化学学报, 2012,28,1223.]doi:10.3866/PKU.WHXB201202234
(17) Lei,G.P.;Liu,C.;Xie,H.Journal of Engineering Thermophysics 2014,35,428.[雷广平,刘 朝,解 辉.工程热物理学报,2014,35,428.]
(18) Bittolo,B.;Valentini,L.;Kenny,J.M.Physica Status Solidi(a) 2010,207,2461.doi:10.1002/pssa.v207:11
(19) Cai,D.;Song,M.;Xu,C.Advanced Materials 2008,20,1706.
(20) Zhu,X.;Ning,G.;Fan,Z.Carbon 2012,50,2764.doi:10.1016/ j.carbon.2012.02.037
(21) Xu,L.;Wei,N.;Zheng,Y.Journal of Materials Chemistry 2012,22,1435.doi:10.1039/c1jm13799a
(22) Novaes,F.D.;Rurali,R.;Ordejon,P.ACS Nano 2010,4, 7596.doi:10.1021/nn102206n
(23) Dimitrakakis,G.K.;Tylianakis,E.;Froudakis,G.E.Nano Letters 2008,8,3166.doi:10.1021/nl801417w
(24) Wu,C.;Fang,T.;Lo,J.International Journal of Hydrogen Energy 2012,37,14211.doi:10.1016/j.ijhydene.2012.07.040
(25) Wesołowski,R.P.;Terzyk,A.P.Physical Chemistry Chemical Physics 2011,13,17027.doi:10.1039/c1cp21590f
(26) Liu,L.;Nicholson,D.;Bhatia,S.K.Chemical Engineering Science 2015,121,268.doi:10.1016/j.ces.2014.07.041
(27) Gupta,A.;Chempath,S.;Sanborn,M.J.;Clark,L.A.;Snurr,R. Q.Molecular Simulation 2003,29,29.doi:10.1080/ 0892702031000065719
(28) Frenkel,S.Understanding Molecular Mimulation—from Algorithms to Applications;Chemical Industry Press:Beijing, 2002;pp 112-113;translated by Wang,W.C.[Frenkel,S.分子模拟—从算法到应用.汪文川译.北京:化学工业出版社,2002:112-113.]
(29) Skarmoutsos,I.;Tamiolakis,G.;Froudakis,G.E.The Journal of Physical Chemistry C 2013,117,19373.
(30) Simon,J.M.;Bellat,J.P.;Salazar,J.M.Molecular Simulation 2014,40,52.doi:10.1080/08927022.2013.845332
Molecular Simulation of Adsorption and Separation Performances for CO2/CH4Mixtures in Graphene/Nanotube Hybrid Structures
LEI Guang-Ping LIU Chao*XIE Hui
(Key Laboratory of Low-Grade Energy Utilization Technologies and Systems of Ministry of Education, Chongqing University,Chongqing 400030,P.R.China)
The adsorption and separation behaviors of CO2and CH4binary mixture in graphene/nanotube hybrid structures(GNHSs)are investigated by grand canonical Monte Carlo(GCMC)combined with molecular dynamics(MD)simulations.CO2is preferentially adsorbed in the adsorbents.Compared with a(6,6)SWCNT (single walled carbon nanotube),GNHSs show improved separation performance.As the temperature rises, the loading of CO2reduces rapidly while the loading of CH4first increases before being reduced.Finally,the kinetic parameters of CO2and CH4,such as self-diffusivity and residence time,are calculated by MD simulation. The CO2molecules diffusing in the GNHS need to overcome a higher barrier relative to that for CH4.The diffusion of the two components in the adsorption layer outside of adsorbent also influences the separation of the mixture.
Graphene/nanotube hybrid structure;Adsorption isotherm;Residence time; Adsorption selectivity;Adsorption coefficient
The project was supported by the National Natural Science Foundation of China(51206195).
国家自然科学基金(51206195)资助项目
O647
10.3866/PKU.WHXB201501291www.whxb.pku.edu.cn
Received:September 24,2014;Revised:January 28,2015;Published on Web:January 29,2015.∗
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10 09:11:20
承德医学院学报(2022年2期)2022-05-23 13:01:36
原子与分子物理学报(2021年1期)2021-03-29 07:28:54
浙江大学学报(工学版)(2016年11期)2016-06-05 09:21:04
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:52
分析测试学报(2015年8期)2016-01-13 06:19:35
合成技术及应用(2015年2期)2016-01-10 10:30:13
原子与分子物理学报(2015年3期)2015-11-24 12:49:39
中国洗涤用品工业(2015年9期)2015-02-28 19:03:04
装备环境工程(2015年4期)2015-02-28 01:20:06
