
阿司匹林固体分散体及其胶囊的制备与体外溶出度研究Δ
2014-12-03 03:06:58杨倪彩胡晨旖朱雅玲林士渭王丽虹管勇军杨群绍兴文理学院元培学院浙江绍兴32000太极集团浙江东方制药有限公司浙江绍兴32000
中国药房 2014年5期
杨倪彩,胡晨旖,朱雅玲,林士渭,王丽虹,管勇军,杨群,2#(.绍兴文理学院元培学院,浙江绍兴32000;2.太极集团浙江东方制药有限公司,浙江绍兴 32000)
口服制剂由于其稳定性好、生产成本低以及顺应性强等优点一直是药物传递系统的首选。然而对难溶性药物阿司匹林,由于其溶解度低、不易被胃肠道吸收,从而造成生物利用度低;此外,阿司匹林还非常容易水解,主要在胃中吸收,长期服用或剂量过大会引起胃肠道刺激,甚至有出血现象[1]。固体分散体(Solid dispersion)系指药物以分子、无定形、微晶或胶态等形式均匀分散在固体载体中所形成的分散体系[2]。以水溶性载体制备的固体分散体可以增加难溶性药物的溶解度和溶出速率,促进吸收,提高药物的生物利用度,增强药物疗效[3-5];同时利用载体的包蔽作用,既可降低药物刺激性与毒副作用,又可隔绝空气、光线和水分,提高药物的稳定性[6]。有研究报道固体分散体多采用传统的熔融法、溶剂法或溶剂-熔融法制备[7-8],制得的固体分散物为致密块状物,粉碎困难,粉碎过程中易造成药物分散状态的改变。喷雾干燥法的干燥温度低,制得的固体分散体为疏松粉体,具有不需粉碎、药物分散度高、溶出快等特点,适合改善难溶性药物的溶出度。因此,本研究以水溶性载体材料聚乙烯吡咯烷酮(PVP K30)为载体,采用喷雾干燥法制备阿司匹林固体分散体,再将固体分散体制成胶囊,以提高阿司匹林的溶出度,提高生物利用度,减少阿司匹林对胃肠道的刺激性,提高药物稳定性。
1 材料
1.1 仪器
AR2130型电子天平[梅特勒-托利多仪器(上海)有限公司];UV-2102PCS型紫外-可见分光光度计[尤尼柯(上海)仪器有限公司];ZRC-8ST溶出仪(天津创兴电子设备制造有限公司);JSM-840型扫描电子显微镜(日本JEOL公司);D/MAX2500型X射线衍射仪(日本理学公司);GP-01粉末振实密度计(厦门市群隆仪器有限公司);JW-04动态比表面快速测定仪(北京精微高博科学技术有限公司)。
1.2 药品与试剂
PVP K30(博爱新开源制药股份有限公司,批号:P130104006-1);阿司匹林对照品(批号:100113-200603,纯度:99.6%)、水杨酸对照品(批号:100106-200303,纯度:≥98%)均购自中国食品药品检定研究院;阿司匹林原料药(山东新华制药股份有限公司,批号:111217,药用级);空心胶囊、滑石粉、硬脂酸镁、微分硅胶均为药用级;浓盐酸、无水乙醇等均为分析纯。
2 方法与结果
2.1 固体分散体的制备
分别按1∶1、1∶2、1∶3、1∶4、1∶5、1∶6、1∶7、1∶8、1∶9、1∶10质量比称取阿司匹林原料药与PVP K3020 g共10份,各加入适量无水乙醇,超声使其溶解,得阿司匹林-PVP K30混合液。将混合液用喷雾干燥仪进行喷雾干燥(操作参数:进风温度120℃,喷雾速度20 ml/min,喷雾压力0.5 mPa,出风温度70~80℃),取出,置干燥器内冷却至室温,即得固体分散体,记为SD1∶1、SD1∶2、SD1∶3、SD1∶4、SD1∶5、SD1∶6、SD1∶7、SD1∶8、SD1∶9、SD1∶10,置干燥器中保存,备用。
2.2 物理混合物的制备
按“2.1”项下质量比称取阿司匹林与PVP K30,研匀,置干燥器中保存,即为阿司匹林与PVP K30的物理混合物,分别记为PM1∶1、PM1∶2、PM1∶3、PM1∶4、PM1∶5、PM1∶6、PM1∶7、PM1∶8、PM1∶9、PM1∶10,备用。
2.3 建立分析方法
2.3.1 检测波长的选择。精密称取阿司匹林对照品5 mg,加入稀盐酸溶液(24→1000),超声20 min使其充分溶解,放至室温后再用稀盐酸溶液稀释定容至100 ml;精密量取10 ml,置于100 ml量瓶中,加稀盐酸溶液稀释至刻度,得阿司匹林对照品溶液。同法配制PVP K30溶液和水杨酸溶液。再精密量取水杨酸溶液5 ml,置于10 ml量瓶中,加稀盐酸溶液稀释至刻度,得水杨酸对照品溶液。取阿司匹林对照品溶液、水杨酸对照品溶液及PVP K30溶液在250~400 nm波长范围内分别进行紫外扫描。结果阿司匹林在277 nm波长处有最大吸收,水杨酸在302 nm波长处有最大吸收,而载体PVP K30在此二波长处均无吸收,故选择277、302 nm分别作为阿司匹林、水杨酸的检测波长。紫外吸收光谱图见图1。

图1 紫外吸收光谱图Fig 1 UV absorbance spectra
2.3.2 标准曲线的绘制。精密称取阿司匹林对照品10 mg,置于100 ml量瓶中,加稀盐酸溶液适量,超声20 min使其充分溶解,放至室温后,加稀盐酸溶液稀释至刻度,摇匀,分别精密量取2、4、6、8、10 ml,置于10 ml量瓶中,加稀盐酸溶液稀释至刻度,摇匀,在277 nm波长处测定吸光度;以吸光度(A)为纵坐标、质量浓度(c)为横坐标进行回归分析,得回归方程A=7.1200c+0.0074(r=0.9999);结果阿司匹林检测质量浓度的线性范围为0.02~0.1 mg/ml。精密称取水杨酸对照品15 mg,同上法溶解制成水杨酸溶液,分别精密量取0.5、1、2、3、5、8、10 ml,置于50 ml量瓶中,加稀盐酸溶液稀释至刻度,摇匀,在302 nm波长处测定吸收度;以吸光度(A)为纵坐标、质量浓度(c)为横坐标进行回归分析,得回归方程A=0.04209c+0.00107(r=0.9999);结果水杨酸检测质量浓度的线性范围为1.5~30 µg/ml。
2.4 溶出度的测定
精密称取阿司匹林原料药、不同比例的阿司匹林固体分散体及其物理混合物适量(约相当于阿司匹林0.1 g),照2010年版《中国药典》溶出度测定法第二法[9],以稀盐酸溶液1000 ml为溶出介质,转速为100 r/min,温度为(37±0.5)℃,依法操作。在5、10、15、30、45、60 min时取样,每次10 ml(即时补充同温溶出介质10 ml),经0.45µm微孔滤膜过滤。取续滤液8 ml,以溶出介质为空白,分别在277、302 nm波长处分别测定阿司匹林和水杨酸的吸光度,代入回归方程计算其各时间点的含量;再将水杨酸含量乘以1.304后,与阿司匹林含量相加即得溶出量,计算药物的累积溶出度,结果见表1和表2。
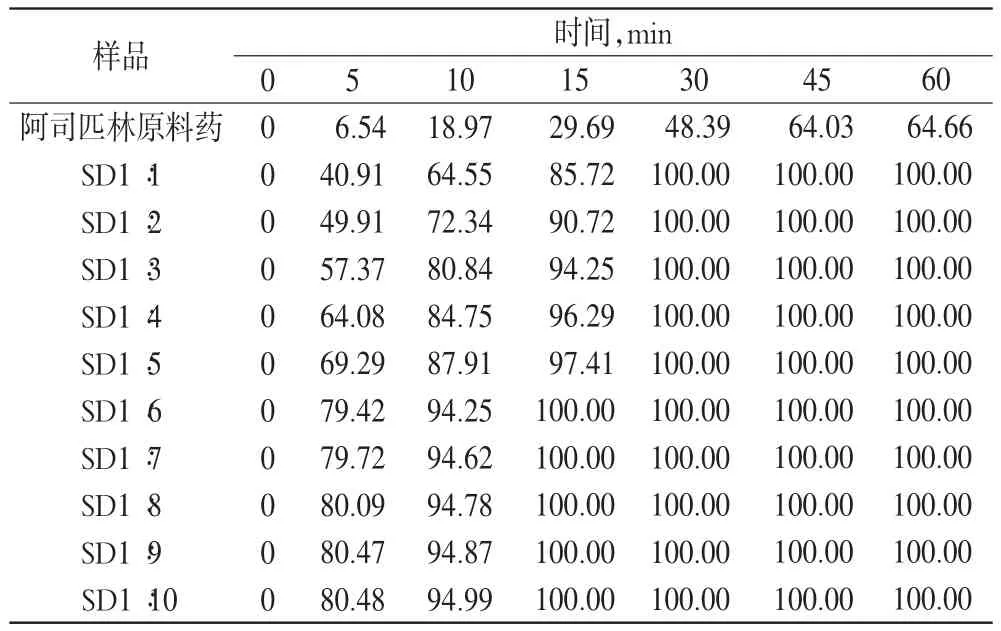
表1 不同比例的阿司匹林固体分散体的体外累积溶出度(%%)Tab 1 The cumulative release in vitro of aspirin solid dispersions with diffevent proportions(%%)
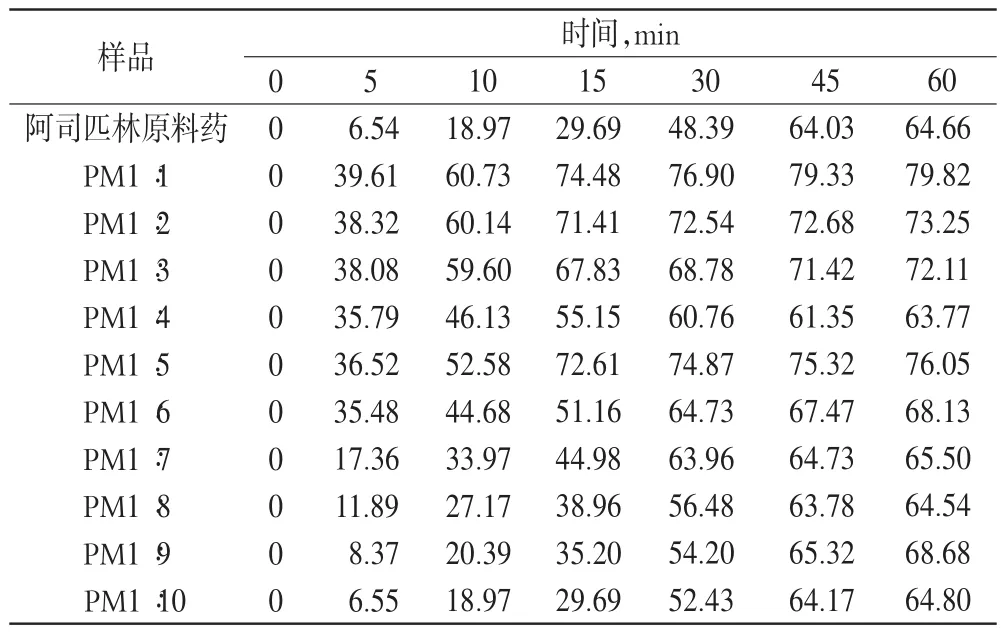
表2 不同比例的阿司匹林物理混合物的体外累积溶出度(%%)Tab 2 The cumulative release in vitro of aspirin physical mixture with diffevent proportions(%%)
由表1和表2可以看出,各比例物理混合物的累积溶出度高于阿司匹林原料药,但增加并不显著。各比例固体分散体累积溶出度均明显高于阿司匹林原料药和物理混合物,30 min时即溶出完全,且累积溶出度与载体材料比例呈正相关;但当质量比达1∶6以后累积溶出度增加不再明显,故一味加大载体比例不可取。通过结果分析显示,固体分散体中阿司匹林与PVP K30的比例以1∶6较为合理。
2.5 固体分散体的表征
2.5.1 粉末X-射线衍射分析。将阿司匹林原料药、PVP K30和各比例的固体分散体及物理混合物分别进行X-射线衍射分析,得到X衍射图谱。工作条件:Cu-Ka,管电流40 mA,高压强度40 kV,衍射角2θ,扫描步距0.026 °,测定范围5~50 °,连续扫描。X-射线衍射结果见图2。
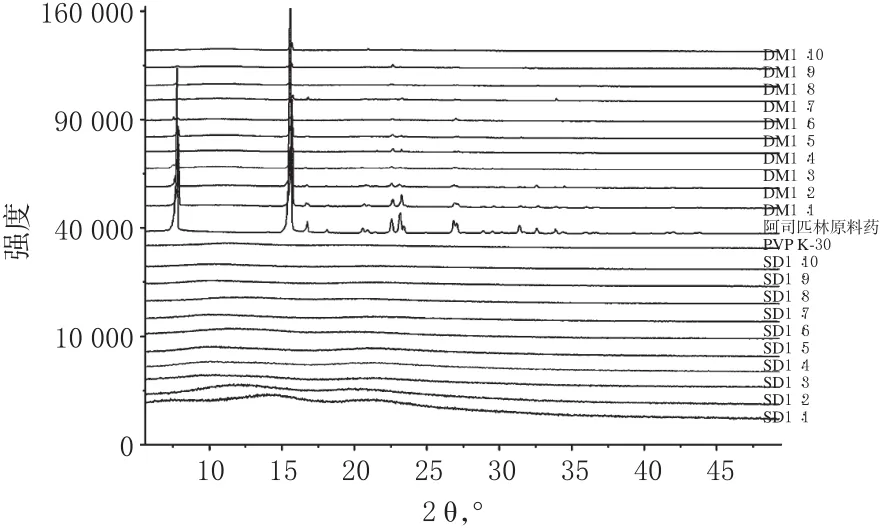
图2 X-射线衍射图Fig 2 X-ray diffraction pattern
由图2可以看出,阿司匹林在7.73、15.56、16.74、18.11、20.58、22.64、23.15、27.04、28.84、29.50、31.34、32.54、33.83 °等位置显示出较强的晶体衍射特征峰,表明阿司匹林以高结晶态存在;PVP K30为无定形粉末,在5~50°无特征结晶衍射峰;在各比例阿司匹林物理混合物中,峰形、峰位与阿司匹林基本一致,峰强度随载体比例增加而减弱;各比例的固体分散体中阿司匹林的晶体衍射峰均消失,推测阿司匹林分子可能进入PVP K-30分子的网状骨架中,被载体包含使药物结晶受到抑制,从而转化为无定形态或分子状态,因此无结晶衍射峰,药物溶出需要的能量少,从而提高了药物的累积溶出度。
2.5.2 扫描电镜分析。取阿司匹林原料药、PVP K30、SD1∶1、SD1∶5、SD1∶10、PM1∶1、PM1∶5、PM1∶10,分别固定于样品台上,用扫描电子显微镜进行验证,观察并记录样品图像,结果见图3。
由图3可以看出,阿司匹林呈针状或柱状结晶,PVP K30呈不规则的无定形物;物理混合物中,阿司匹林以结晶形式分散在载体材料PVP K30中,只是随着载体材料比例增加,稀释了药物的晶体密度;固体分散体中,阿司匹林的结晶均消失,固体分散体呈不规则无定形物,表明阿司匹林以喷雾干燥法制备成固体分散体后,阿司匹林由晶体转变成无定形态或分子状态分散,药物溶出需要的能量少,这可能是其体外溶出度提高的一个重要原因。
2.5.3 比表面积的测定。将待测样品于29℃下脱气30 min,用氮气吸附法,采用动态比表面快速测定仪测定并记录SD1∶6的比表面积。结果显示阿司匹林制成固体分散体后,比表面积约为原料药的4.2倍。根据Noyes-Whitney方程:dc/dt=kS(cs-c)(式中dc/dt表示药物的溶出速率,k表示溶出速率常数,S表示溶出质点暴露于介质的表面积,cs为药物的溶解度),在漏槽条件下,c→0,dc/dt=kScs,即dc/dt与S呈正比。由结果可知,阿司匹林制成固体分散体后,比表面积提高了3.2倍,这应该是固体分散体中药物溶出度提高的另一个重要原因。
2.6 阿司匹林固体分散体胶囊的制备
2.6.1 润滑剂的选择。取SD1∶6,过18目筛整粒,分别向其中加入1%的滑石粉、微粉硅胶、硬脂酸镁作为润滑剂,混合均匀,按注入法[10]测定并计算休止角(tanθ=h/r),比较不同润滑剂的助流作用。结果以微粉硅胶为润滑剂所得休止角最小,故确定以微粉硅胶为润滑剂。再分别向SD1∶6加入0.1%、0.2%、0.3%、0.4%、0.5%、0.8%、1.0%微粉硅胶,以粉末振实密度计轻敲后测定每次轻敲后体积减少度C,代入川北方程:n/C=n/a+1/ab,计算最终体积减少度a和充填速度常数b(a越小,振动和压缩流动越好;b反映粉体的充填特性,b越大,充填速度越大,充填越容易进行)。以a、b为指标,确定最佳微粉硅胶的量。结果以加入0.2%微粉硅胶的SD1∶6的a最小(为0.1),b最大(为1.3),2%微粉硅胶为最佳润滑剂量。
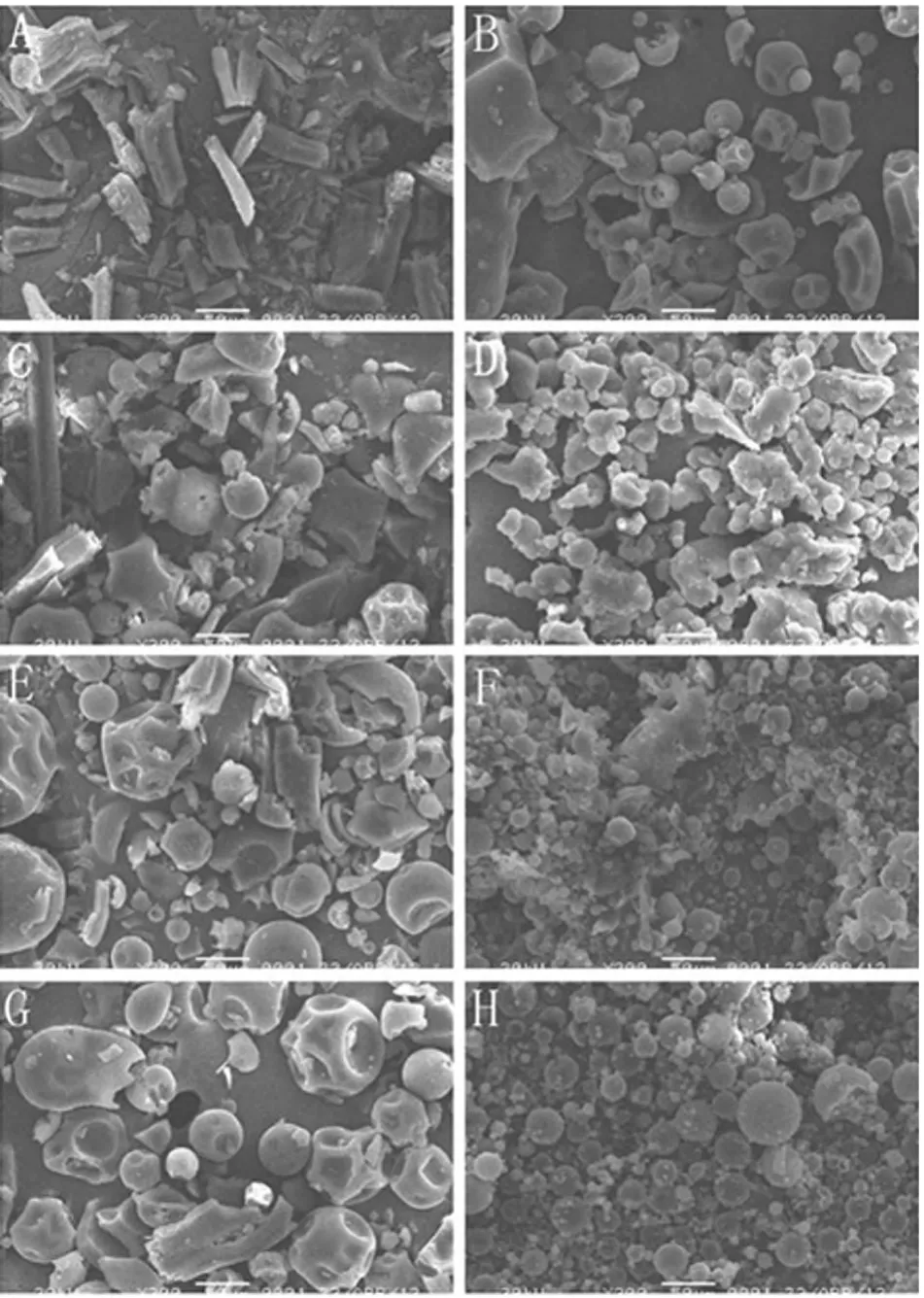
图3 扫描电镜图(×300)A.阿司匹林原料药;B.PVP K30;C.PM1 ∶1;D.SD1∶1;E.PM1∶5;F.SD1∶5;G.PM1∶10;H.SD1∶10Fig 3Scanning electron microscop(e×300)A.aspirin crude drug;B.PVP K30;C.PM1 ∶1;D.SD1 ∶1;E.PM1 ∶5;F.SD1∶5;G.PM1∶10;H.SD1∶10
2.6.2 胶囊的制备。取SD1∶6,过18目筛,加入0.2%的微粉硅胶,混合均匀,用胶囊板填充胶囊。检测胶囊装量差异[9]、溶出度[9]。结果平均装量为0.292 g,装量差异为±1.9%,符合《中国药典》标准,30 min时阿司匹林累积溶出度达99.8%。
3 讨论
本试验选择喷雾干燥法制备阿司匹林固体分散体,有其独特的优势。传统溶剂法、熔融法、溶剂-熔融法制得的溶液浓缩过程中黏度较高,导致溶剂难以完全挥干、干燥温度较高、干燥物结构致密、粘壁严重,收率较低。而喷雾干燥法具有:(1)避免高温操作,保护热敏性药物;(2)喷出雾滴瞬间干燥,保证了药物的高度分散性,增大制剂的比表面积,从而能更大程度地改善其溶出;(3)工艺重现性好,药物溶出快,分散体粒子无需粉碎即可满足各类固体制剂的制备要求,产量大,利于工业化大生产,是一种较理想的固体分散体的制备方法。
体外溶出度测定结果表明,固体分散体中药物的溶出度显著优于其物理混合物及原料药,推断除了载体对药物的润湿和增溶作用外,分散度的增加是主要的促溶机制[10],为提高其口服生物利用度提供了必要的条件。
X-射线衍射和扫描电镜的结果表明,固体分散体中PVP K30与阿司匹林可能由于形成了复合物或氢键等相互作用而抑制药物晶核形成和结晶的生长,可使药物以无定形或分子状态存在于固体分散体中,由于无晶格束缚,自由能大,相比稳定晶型具有较高的溶解度,从而提高溶出速度。
比表面积测定结果表明,药物制成固体分散体后,比表面积增加了3.2倍,从另一方面解释了药物溶出增加的原因。
综上,制备的阿司匹林固体分散体胶囊处方、工艺简单易行,质量达到了设计要求,有利于提高药物溶出速度、掩盖药物的苦味,利于口服,可为工业化生产提供依据。
[1]新怡康.普通阿司匹林、阿司匹林肠溶片临床应用胃肠道不良反应的比较[J].临床和实验医学杂志,2006,5(8):1201.
[2]Desai J,Alexander K,Riga A.Characterization of polymeric dispersions of dimenhydrinate in ethyl cellulose for controlled release[J].Int J Phan,2006,308(1/2):115.
[3]朱盛山.药物新剂型[M].北京:化学工业出版社,2003:31.
[4]娄树立,吴丽伟.论阿司匹林的临床应用[J].黑龙江医药,2010,23(2):255.
[5]Redondo S,Santos-Gallego CG,Ganado P,et al.Acetylsalicylic acid inhibits cell proliferation by involving transforming growth factor-β[J].Circulation,2003,107(4):626.
[6]崔福德.药剂学[M].7版.北京:人民卫生出版社,2011:347-348.
[7]邹阳,黄华.固体分散法制剂工艺研究进展[J].中国医药工业杂志,2005,36(10):648.
[8]陆彬.药物新剂型与新技术[M].北京:人民卫生出版社,1998:3.
[9]国家药典委员会.中华人民共和国药典:二部[S].2010年版.北京:中国医药科技出版社,2010:附录8、85、385.
[10]崔福德.药剂学实验指导[M].北京:人民卫生出版社,2010:125-352.
猜你喜欢
陶瓷学报(2020年5期)2020-11-09 09:23:00
国防交通工程与技术(2020年2期)2020-05-25 06:46:16
中成药(2018年1期)2018-02-02 07:19:55
中国资源综合利用(2017年2期)2018-01-22 02:45:12
中成药(2017年10期)2017-11-16 00:50:08
中成药(2017年10期)2017-11-16 00:50:03
数理化解题研究(2017年2期)2017-04-13 07:40:16
特产研究(2016年3期)2016-04-12 07:16:16
材料科学与工程学报(2016年5期)2016-02-27 07:11:31
中国教育技术装备(2015年6期)2015-03-01 02:37:08
