
无模板两步法合成亚微米A 型沸石
2014-11-25 09:54:28刘艳娜孙彦琳
硅酸盐学报 2014年10期
刘艳娜,白 璞,龙 丽,简 攀,肖 松,孙彦琳
(1.昆明理工大学化学工程学院,昆明 650500;2.昆明理工大学冶金与能源工程学院,昆明 650093)
沸石分子筛是一类多孔晶体材料,孔道尺寸一般为0.3~3nm,其特殊的微孔结构使其具有很多重要的性质,诸如离子交换性[1]、催化活性[2]、吸附性[3]等。一般认为,晶粒尺寸越小的沸石具有更大的外比表面积、更短的扩散距离以及更多暴露的活性中心[4],因此对上述性质均有一定的促进作用。A 型沸石由于凝胶组成简单、拓扑结构规整、晶化速度快、应用性广泛,经常被用来当做研究沸石晶化过程的典型实例。目前合成小粒径A 型沸石的主要手段包括:使用有机模板剂[5]、降低晶化温度[6]、延长陈化时间[7]等。在A 型沸石合成过程中加入有机模板剂四甲基氢氧化铵(TMAOH)可以极大促进其成核过程,制得产物晶粒尺寸可达100nm 左右,且晶型良好,是目前实验室范围制备纳米A 沸石最常用的方法[5,8]。然而由于在合成过程中需要大量使用较昂贵的TMAOH,后续煅烧除去模板剂打开孔道需要消耗大量的能源,且存在产率低、易团聚等缺点限制了其应用。降低晶化温度可以更容易控制晶体的生长速度,从而获得粒径较小的晶体,但此法制备出来的晶型一般不是很好,且合成周期也过长[6--7]。因此,无模板剂短周期内合成粒度分布均一且晶型较好的亚微米A 型沸石成为研究的热点。
大量研究表明沸石的陈化过程不仅导致硅铝凝胶组成与结构的变化,而且最终将影响晶化产物的晶粒尺寸、形貌及结构[4,9]。李守贵等[10]应用核磁共振和Raman光谱研究了L 型沸石初始凝胶的陈化机制,发现在不同的陈化时间,体系中硅铝酸根离子状态发生了明显的变化,具体表现为高聚态硅酸根离子发生了解聚,Al(OH)-4对应的特征峰消失。熊晓云等[11]应用高效NaY 沸石导向剂为硅源快速合成 了超细A 型 沸 石,29Si NMR 和UV Raman 研究表明高效NaY 沸石导向剂中存在大量的六元环等低分子量硅铝酸根前驱体,它们的存在有利于A型沸石的成核及生长,实验中所用的导向剂为经过25 ℃陈化24 h 的NaY 沸石硅铝酸盐凝胶。Valtchev等[6]不添加有机模板剂配制初始凝胶,然后经过室温振荡陈化反应10d得到了平均晶粒尺寸为400~500nm 的A 型沸石,但由于没有经过升温晶化过程,合成出来A 型沸石产物晶型很不规整,周期过长。Alfaro等[7]以硅溶胶为硅源,异丙醇铝为铝源配制初始凝胶,经过144h 室温陈化和100℃搅拌晶化24h制备出粒径约200~500nm 的A 型沸石,此法周期过长,且合成的产物粒度分布不均匀,成本也相对较高。
因此,在前人基础上,设计了经由两步法制备亚微米A 型沸石。第一步中初始凝胶陈化过程是在温度为35℃且在搅拌下完成的;第二步对陈化一定时间的硅铝凝胶进行升温晶化处理。此两步法设计主要基于以下3点:1)低温有利于成核,但温度过低会影响初始硅铝凝胶的溶解过程和导致成核诱导期延长;2)第一步中适当的搅拌有利于硅铝凝胶及晶核的均匀分布,利于合成具有较窄粒度分布范围的产物[4];3)升温晶化使晶化周期有效缩短,且生成产物晶型较好。应用X 射线衍射仪、红外光谱仪、Raman光谱仪、粒度分布仪、X 射线荧光光谱仪、扫描电子显微镜等对第一步中作用不同时间的硅铝凝胶固相及经过第二步得到的最终产物进行检测。通过对检测结果进行分析,对两步法制备亚微米A 型沸石的作用机制进行了初步探讨。
1 实验
1.1 样品制备
按3.165 Na2O∶Al2O3∶1.926SiO2∶128 H2O摩尔配比配制初始硅铝凝胶[12]。具体步骤为向氢氧化钠溶液中缓慢加入NaAlO2,在室温搅拌,配制出溶液记为溶液1。向另外的氢氧化钠溶液中缓慢加入Na2SiO3·9H2O,在室温搅拌,配制出的溶液记为溶液2。将溶液1快速倒入溶液2中,再继续搅拌10min即配制出初始硅铝凝胶,标记为ASG-0。
初始硅铝凝胶经由两步合成目的样品。第一步为陈化:将ASG-0移入35℃水浴中,搅拌陈化一定时间。将经过搅拌陈化23,56,85,139h的硅铝凝胶分别标记为ASG-23,ASG-56,ASG-85,ASG-139。分别取出一定量的ASG-0,ASG-23,ASG-56,ASG-85,ASG-139 进行离心分离,下层固相经过40℃真空干燥完全后检测备用。第二步为晶化:另外再分别取一定量的上述硅铝凝胶装入50mL 反应釜中,100 ℃晶化4h。晶化后的反应液经过超声,抽滤洗涤至中性,110 ℃干燥24h 后即得到A型沸石成品。将用ASG-0,ASG-23,ASG-56,ASG-85,ASG-139 制备出来的成品分别标记为S-0,S-23,S-56,S-85,S-139。表1为合成样品的初始硅铝凝胶摩尔配比及工艺参数。
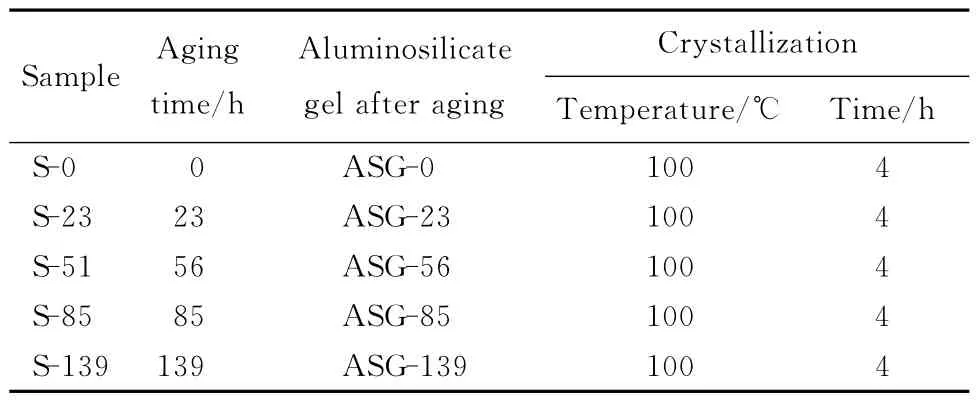
表1 初始凝胶摩尔配比及工艺条件Table 1 Composition of initial aluminosilicate gel and conditions of sample synthesized
1.2 表征
用德国TENSOR27 型Fourier变换红外光谱仪待测量样品的红外吸收光谱,KBr 压片,范围400~4 000cm--1。用英国invia型激光显微Raman光谱仪测量样品的Raman 光谱。用日本Rigaku D/max-2200X型X 射线衍射仪样品的物相,Cu靶Kα辐射,扫描角度为3°~80°。用日本ZSX100e型X 射线荧光光谱仪分析样品的主要元素含量,并对其SiO2/Al2O3进行计算。用LS603型激光衍射粒度分析仪(珠海欧美克科技有限公司)测定样品的粒度分布,D50表示从粒径分布曲线中确定的累计体积达到50%时的产物粒径大小。用荷兰FEIQuanta 200型扫描电子显微镜对样品进行观察,获得有关样品形貌及晶粒大小、分布等信息。
2 结果与讨论
2.1 物相分析
图1a为硅铝凝胶固相的XRD 谱,图1b为硅铝凝胶经过晶化后得到产物的XRD 谱。由图1a可知,硅铝凝胶在35℃搅拌下分别陈化0、23、56h其对应固相均为无定形态。当延长陈化时间至85h时开始出现较微弱的A 型沸石特征峰,说明在小于85h这段陈化时间内体系是发生变化的,主要体现为诱导A 型沸石晶核的形成,而只有当晶核积累至一定程度时其衍射峰才能被检测到[13]。继续延长陈化时间至139h,A 型沸石的特征峰强度明显增强,说明这段时期体系的变化已经由晶核的形成变为晶体的生长。由图1b可知经不同陈化时间的硅铝凝胶晶化后得到产物均为A 型沸石,且随着陈化时间的延长对应产物衍射峰强度呈逐渐下降趋势,说明随着陈化时间的延长合成的A 型沸石产物相对结晶度随之下降。
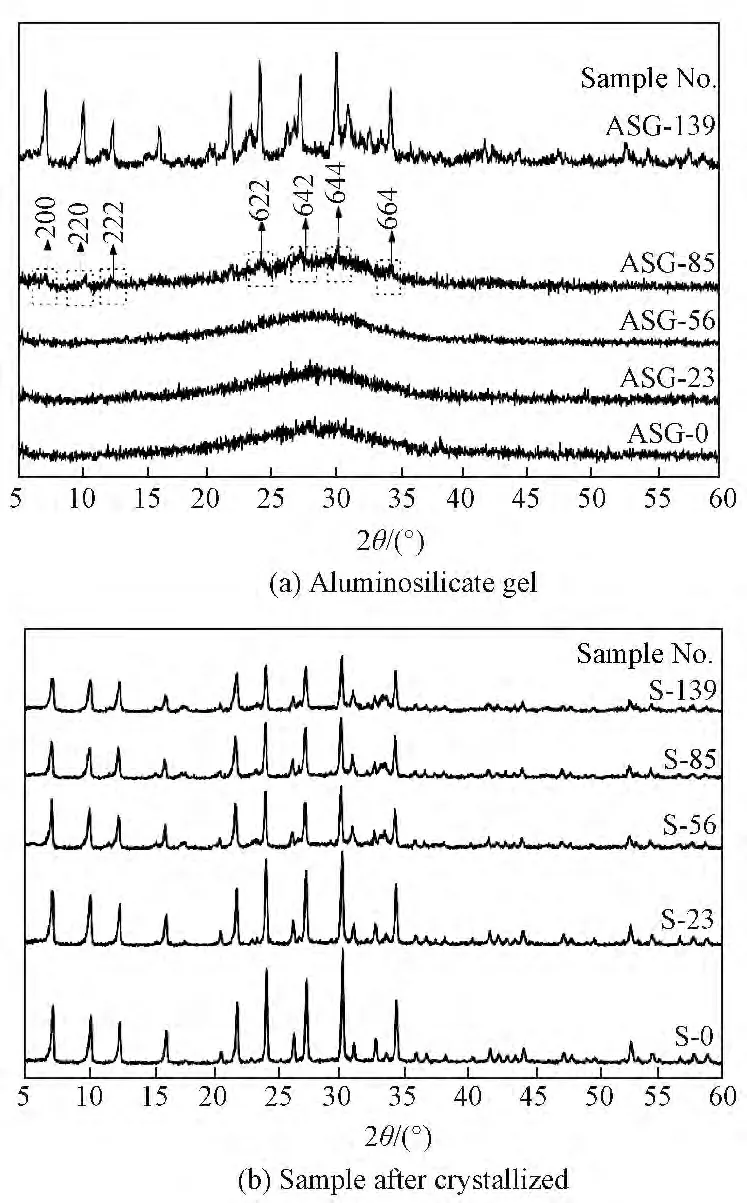
图1 不同陈化时间硅铝凝胶固相及经过晶化后得到产物的XRD 谱Fig.1 XRD patterns of the aluminosilicate gel solid phase with different aging time and the as-synthesized samples after crystallized
2.2 光谱分析
图2为硅铝凝胶固相红外光谱,其中995cm--1附近为T—O—T 键不对称伸缩振动;710cm--1左右为T—O—T 键的对称伸缩振动;560cm--1附近为双环结构单元振动;460cm--1附近为TO4弯曲振动[14]。由图2 可以看出,随着陈化时间的延长,995、710、460cm--1附近处的特征峰强度逐渐增强;560cm--1附近处的特征峰对应的是骨架中的双四元环(D4R)结构,其在陈化时间增至56h时开始显现出来,随后逐渐增强,特别是在85~139h 明显增强。D4R 是组成A 型沸石最重要的次级单元结构之一[15],其特征峰的出现和增强反映出A 型沸石逐渐形成和生长的过程。
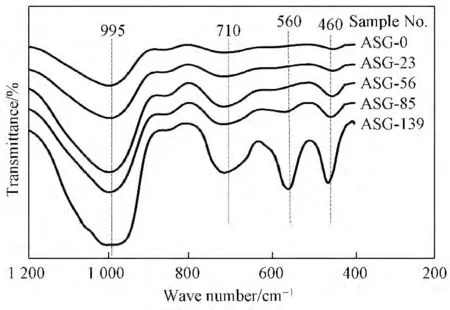
图2 硅铝凝胶固相红外光谱Fig.2 IR spectra of the aluminosilicate gel solid phase
图3为陈化时间不同的硅铝凝胶固相Raman光谱。由图3可知,初始硅铝凝胶分别在508、579、726、857、1 002cm--1处出现了较微弱的特征峰。随着陈化时间的延长,508cm--1处的特征峰逐渐向左偏移,且强度逐渐增强;579cm--1处的特征峰逐渐消失;726cm--1附近的特征峰强度逐渐增强。当陈化时间延长至85h时,在341、751、1 099cm--1处出现新的特征峰,这些新出现的特征峰都归属于A 型沸石的Raman特征峰[16],说明此时已经A 型沸石已经开始形成。陈化时间延长至139h 时,在342、411、500、721、749、850、982、1 038、1 099cm--1处对应的较明显的特征峰均属于A 型沸石的特征峰[16],说明A 型沸石在85~139h这段区间已经开始生长。此结果与红外及XRD 结果一致,但相对于前2种检测,Raman光谱反映出了过程中硅铝凝胶固相更细微的变化。研究表明,500~530cm--1附近对应的是初级单元结构四元环[17],在A 型沸石的形成和生长过程中,四元环扮演着重要的角色,其不仅组成了D4R 而且也是构成α 笼、β笼的重要物种[15]。由图3可见四元环结构对应特征峰强度都较明显,说明其在整个A 型沸石的形成过程中占主导地位。
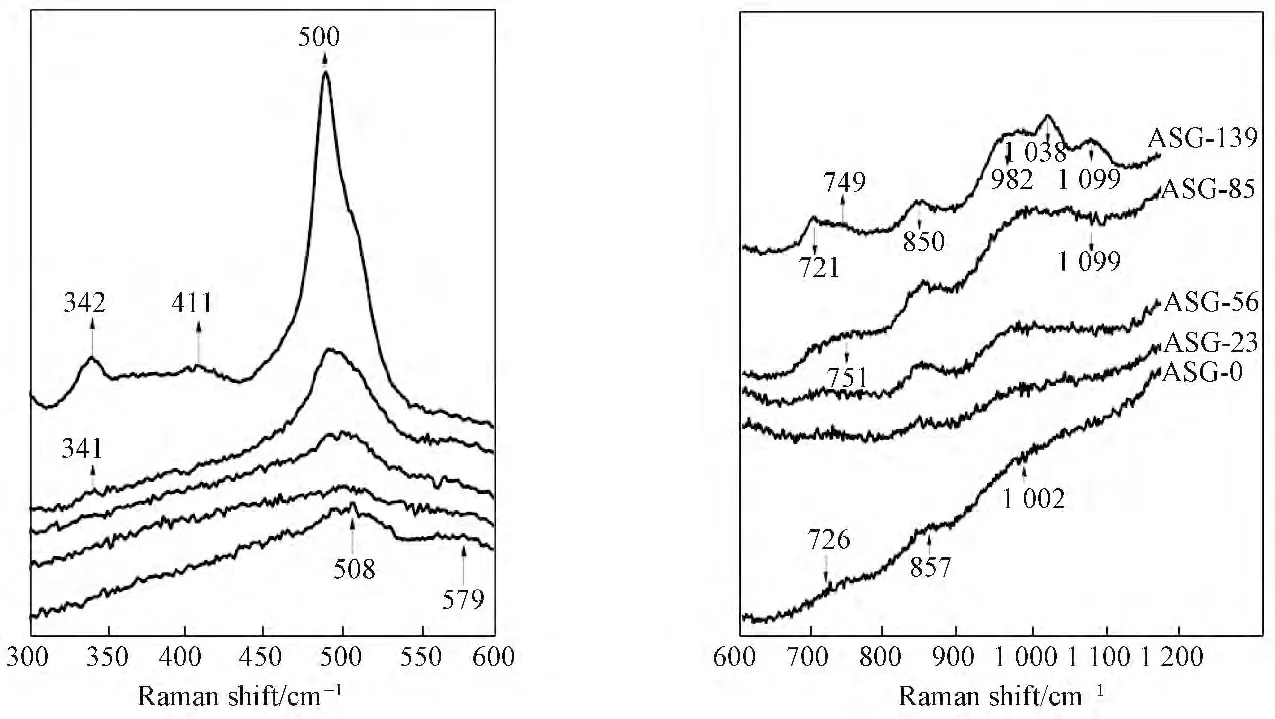
图3 硅铝凝胶固相Raman光谱Fig.3 Raman spectra of the aluminosilicate gel solid phase
2.3 硅铝凝胶固相SiO2/Al2O3变化分析
图4为硅铝凝胶固相SiO2/Al2O3摩尔比随陈化时间变化关系。由图4可知:硅铝凝胶固相中摩尔比n(SiO2)/n(Al2O3)随着陈化时间的延长呈逐渐下降的趋势,其中在0~23h及85~139h时间段下降较明显。根据A 型沸石的液相生长机理,硅铝凝胶溶解进入溶液,在液相中硅酸根离子与铝酸根离子在Na+作用下形成A 型沸石晶核。此时的晶核骨架具有较高的硅铝比[18],这就导致了在陈化的初始阶段,硅铝凝胶固相中大量的硅酸根离子开始溶解进入液相,从而导致了此时间段n(SiO2)/n(Al2O3)明显下降。由XRD 结果可知在85~139h时晶体显著生长(图1a),生成的A 型沸石晶体骨架中n(SiO2)/n(Al2O3)相对较小(等于或略大于2),溶液中的硅源与铝源不断累积进入固相,进而导致这一时间段硅铝凝胶固相中n(SiO2)/n(Al2O3)的较明显下降。
2.4 粒度变化分析
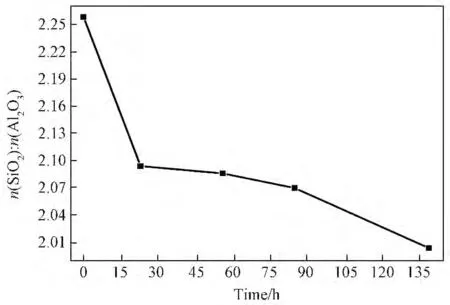
图4 经不同陈化时间的硅铝凝胶固相的n(SiO2)/n(Al2O3)比值Fig.4 Ratio of n(SiO2)/n(Al2O3)the aluminosilicate gel solid phase with different aging time
图5a为不同陈化时间对应的硅铝凝胶固相的粒度分布图。由图5a可知:随着陈化时间的延长得到的硅铝凝胶固相粒度分布范围逐渐变窄,平均粒径也随之减小。图5b为不同陈化时间的硅铝凝胶经过晶化后得到产物的粒度分布图。由图5b可知:在0~85h时,随着陈化时间的延长,经过晶化后的产物粒度分布范围也逐渐变窄,但当陈化时间由85h增至139h时,所得产物粒度分布范围变宽,平均粒径也随之变大。图6为硅铝凝胶及合成产物的平均粒径随陈化时间变化关系图。由图6可知,随着陈化时间的延长其相应的硅铝凝胶固相平均粒径由初始ASG-0对应的11.6μm 减小至ASG-139对应的1.02μm,这说明随着陈化时间的延长,初始硅铝凝胶逐渐溶解,而溶解后的硅铝凝胶为A 型沸石成核及生长提供营养,这验证了成长过程的液相转变机制[9]。液相转变机制认为,沸石晶核的形成发生在液相中或在硅铝凝胶与液相界面[19]。陈化过程中搅拌步骤不仅加快了硅铝凝胶的溶解速率,同时也促进了形成的A 型沸石晶核从凝胶表面释放到液相以及体系和晶核的均匀分布。随后的85~139h这段时期既有A 型沸石晶核的生长同时也有硅铝凝胶继续溶解为晶核的生长提供营养[20],但由于温度过低(35℃),晶核生长十分缓慢,因此在这段时期被测物粒度在总体上仍表现为减小的趋势。相应地,经过升温晶化后产物的平均粒径由S-0对应的2.46μm 减小至S-85对应的0.58μm,这主要是由于随着陈化时间的延长导致生成了更多的晶核,而在升温晶化过程中,晶核数量越多越有利于生成粒径较小的晶体[21,22]。然而,继续延长陈化时间至139h时对应产物S-139平均粒径增至2.47μm,造成这种现象的具体原因将结合SEM 照片进行分析。由图6还可以看出:分别经历陈化时间0、23、56、85h的硅铝凝胶在经过升温晶化后,体系中固相物质的平均粒径在较短时间(4h)内均出现较大幅度的下降,这说明升温晶化过程极大的促进了硅铝凝胶的溶解,并且大大缩短了A 型沸石的成核诱导期和生长期。
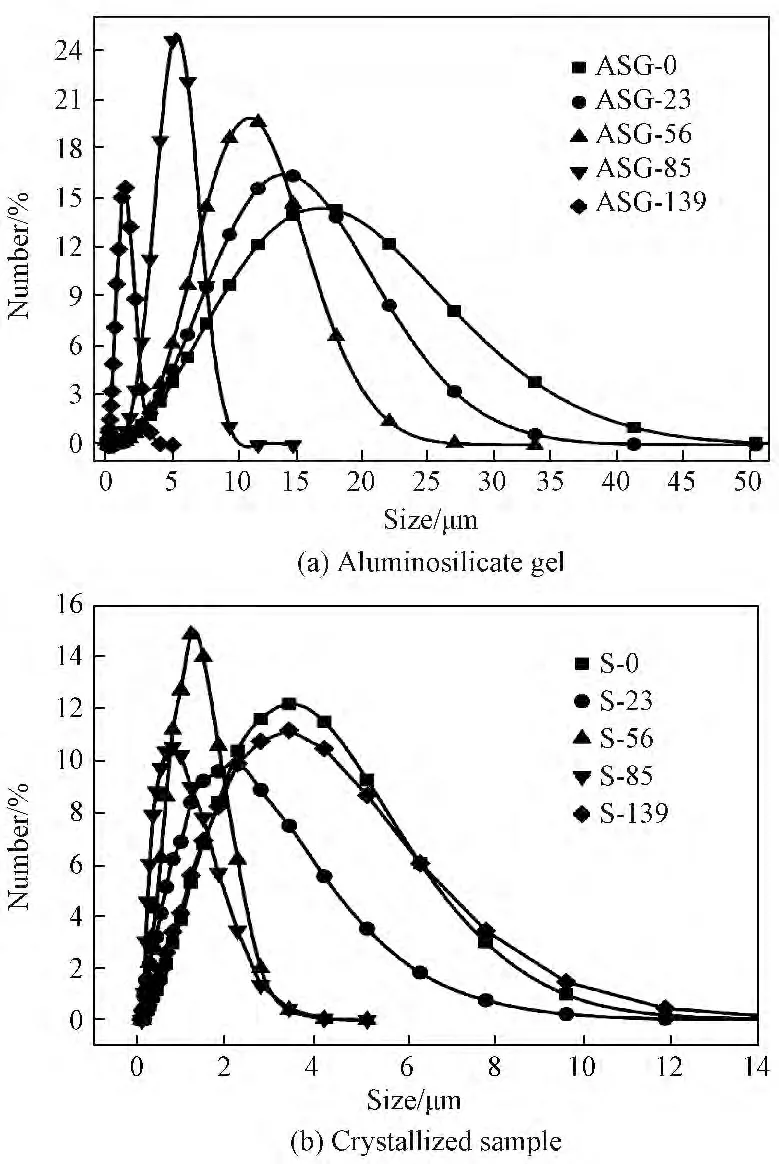
图5 不同陈化时间硅铝凝胶固相及经过晶化后得到产物的粒度分布图Fig.5 Particle size distributions of the aluminosilicate gel solid phase with different aging time and the crystallized samples
2.5 显微结构分析
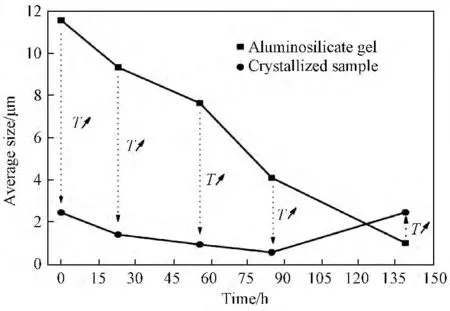
图6 硅铝凝胶固相及合成产物的平均粒径与陈化时间(T)之间的关系Fig.6 Average size of the aluminosilicate gel solid phase and the as-synthesized samples with different aging time(T)
ASG-139的SEM 照片如图7a所示。由图7a可见:此时已经有A 型沸石特有的立方体结构开始生成,但形貌不规则,且有大量无定型形貌的物质的存在,这与XRD 检测结果一致。晶化样品的SEM 照片如图7b~图7f所示。由图7b~图7e可知,随着陈化时间的延长,合成的立方形结构产物粒径逐渐变小,其中样品S-85 粒径最小(约500nm),且粒径分布十分均匀。而当陈化时间延至139h时,对应样品S-139 出现团聚现象,组成团聚体颗粒的基本粒子尺寸与样品S-85 相似,但形貌变的不规整。这说明陈化时间过长反而对A型沸石的形成不利,且不利于生成粒径分布较窄、分散较好的亚微米A 型沸石,此结果与粒度分布结果一致。
由于小粒径沸石对X 射线的消光效应使其相应XRD 峰呈宽化趋势[23],且随着陈化时间的延长,得到的A 型沸石产物粒径越小,此现象越明显,对应的相对结晶度也随之下降(图1b),而并不能说明得到的A 型沸石产物的真实结晶度下降。前面已经分析了样品S1作用时间过长对A 型沸石形成是不利的,这与小粒径沸石的消光效应一起导致了样品S-139表观结晶度最低。
2.6 两步法合成亚微米A 型沸石合成机制
初始的无定形硅铝凝胶经陈化后,随着陈化时间延长,无定形硅铝凝胶颗粒逐渐溶解减小(图6)。硅铝凝胶溶解生成的铝酸根离子及硅酸根离子在溶液中发生一系列复杂的解聚、缩聚、自组装等过程生成形成A 型沸石所必需的一些结构单元,例如四元环(图3)、D4R(图2)等。低温有利于成核,而限制晶体生长,这些结构单元通过自组装过程形成A 型沸石的晶核,且随着陈化时间的延长,生成的晶核数量随之增加(图1a)。晶化温度(100℃)相对较高,温度升高更有利于晶体的生长,硅铝凝胶在高温下溶解后直接为晶核的生长提供原料,此时体系中存在的晶核数量越多则生成的A 型沸石产物粒径越小(图6,图7)。

图7 硅铝凝胶固相(ASG--139)和晶化样品的SEM 照片Fig.7 SEM images of ASG--139and crystallized samples
3 结论
在无模板剂存在下,经由两步法成功合成粒径分布均匀(平均粒径约500nm)且晶型较好的A 型沸石,其粒径明显小于不经过第一步作用得到产物的粒径(2.47μm)。硅铝凝胶固相随着第1步作用时间的延长其尺寸、结构、组成均发生了较大的变化,这些变化促进了更多分布均匀的A 型沸石晶核的形成,相应的经过第2步得到的A 型沸石产物粒径也随之减小,但第一步作用时间过长则不利于形成分散性好、形貌规整的A 型沸石产物。此外,验证了A 型沸石的液相转变机理,同时对合成其他类型亚微米沸石具有一定指导意义。
[1]MERCEILLE A,WEINZAEPFEL E,BARRÉY,et al.The sorption behaviour of synthetic sodium nonatitanate and zeolite A for removing radioactive strontium from aqueous wastes[J].Sep Purif Technol,2012,96:81-88.
[2]张敏,肖质文,何红运.Cu-β 沸石的合成、表征及催化性能[J].无机化学学报,2011,27(3):427-433.ZHANG Min,XIAO Zhiwen,HE Hongyun.Chin J Inorg Chem(in Chinese),2011,27(3):427-433.
[3]WHITE J C,DUTTA P K,SHQAU K,et al.Synthesis of ultrathin zeolite Y membranes and their application for separation of carbon dioxide and nitrogen gases[J].Langmuir,2010,26(12):10287-10293.
[4]TOSHEVA L,VALTCHEV V P.Nanozeolites:synthesis,crystallization mechanism,and applications[J].Chem Mater,2005,17(10):2494-2513.
[5]MINTOVA S,OLSON N H,VALTCHEV V P,et al.Mechanism of zeolite A nanocrystal growth from colloids at room tem-perature[J].Science,1999,283(5404):958-960.
[6]VALTCHEV V P,TOSHEVA L,BOZHILOV K N.Synthesis of zeolite nanocrystals at room temperature[J].Langmuir,2005,21(23):10724-10729.
[7]ALFARO S,RODRIGUEZ C,VALENZUELA M A,et al.Aging time effect on the synthesis of small crystal LTA zeolites in the absence of organic template[J].Mater Lett,2007,61(24):4655-4658.
[8]FAN W,OGURA M,SANKAR G,et al.In situ small-angle and wide-angle X-ray scattering investigation on nucleation and crystal growth of nanosized zeolite A [J].Chem Mater,2007,19(8):1906-1917.
[9]CUNDY C S,COX C A.The hydrothermal synthesis of zeolites:Precursors,intermediates and reaction mechanism[J].Microporous Mesoporous Mater,2005,82(1):1-78.
[10]熊晓云,范峰滔,马军,等.用高效NaY 沸石导向剂快速合成A 型沸石[J].高等学校化学学报,2007,28(1):21-25.XIONG Xiaoyun,FAN Fengtao,MA Jun,et al.Chem J Chin Univ(in Chinese),2007,28(1):21-25.
[11]李守贵,李锡凯,徐如人.L沸石导向剂陈化机制的研究[J].高等学校化学学报,1992,13(2):145-148.LI Shougui,LI Xikai,XU Ruren.Chem J Chin Univ(in Chinese),1992,13(2):145-148.
[12]YUSOF A M,NIZAM N A,RASHID N A A.Hydrothermal conversion of rice husk ash to faujasite-types and NaAtype of zeolites[J].J Porous Mater,2010,17(1):39-47.
[13]张敏,高丙莹,何红运.新型V-Ni-β沸石的合成、表征及催化性能[J].无机化学学报,2012,28(11):2355-2362.ZHANG Min,GAO Bingying,HE Hongyun.Chin J Inorg Chem(in Chinese),2012,28(11):2355-2362.
[14]YAO J F,YU L,ZHANG L X,et al.Influence of glycerol cosolvent on the synthesis of size controllable zeolite A [J].Mater Lett,2011,65(14):2304-2306.
[15]REN L M,LI C J,FAN F T,et al.UV-raman and NMR spectroscopic studies on the crystallization of zeolite Aand a new synthetic route[J].Chem Eur J,2011,17(22):6162-6169.
[16]DUTTA P K,SHIEH D C.Crystallization of Zeolite A:A spectroscopic study[J].J Phys Chem,1986,90(11):2331-2334.
[17]FAN F T,FENG Z C,LI G N,et al.In situ UV raman spectroscopic studies on the synthesis mechanism of zeolite X[J].Chem Eur J,2008,14(17):5125-5129.
[18]马淑杰,刘孔凡,崔美珍,等.A 型沸石的生成机理[J].高等学校化学学报,1984,5(2):158-162.MA Shujie,LIU Kongfan,CUI Meizhen,et al.Chem J Chin Univ(in Chinese),1984,5(2):158-162.
[19]王仰东,刘杨,董家禄,等.小晶粒A 型分子筛的合成及其晶貌[J].催化学报,1998,19(6):610-612.WANG Yangdong,LIU Yang,Dong Jialu,et al.Chin J Catal(in Chinese),1998,19(6):610-612.
[20]王建,董家禄,刘杨,等.偏高岭土合成4A 沸石机理的研究[J].无机化学学报,2000,16(1):31-36.WANG Jian,DONG Jialu,LIU Yang,et al.Chin J Inorg Chem(in Chinese),2000,16(1):31-36).
[21]黄先亮,王正宝,无模板剂两步法合成小颗粒ZSM-5沸石团聚体[J].催化学报,2011,32(11):1702-1711.HUANG Xianliang,WANG Zhengbao.Chin J Catal(in Chinese),2011,32(11):1702-1711.
[22]张铨昌.沸石分子筛的合成与应用[J].硅酸盐学报,1992,20(6):544-550.ZHANG Quanchang.J Chin Ceram Soc,1992,20(6):544-550.
[23]ADNADJEVIC B,VUKICEVIC J,FILIPOVIC-ROJKA Z,et al.The influence of NaX zeolite particle size on crystallinity measured by the XRD method[J].Zeolites,1990,10(7):699-702.
猜你喜欢
化工学报(2022年7期)2022-08-10 09:49:42
陶瓷(2021年5期)2021-06-29 08:07:16
Acta Mathematica Scientia(English Series)(2021年1期)2021-04-08 12:52:22
四川水泥(2020年10期)2020-10-27 06:34:12
中国茶叶加工(2020年3期)2020-10-21 08:06:48
陶瓷学报(2019年6期)2019-10-27 01:18:38
山东化工(2018年10期)2018-06-07 04:33:33
石油化工应用(2017年10期)2017-11-08 03:20:06
科学导报·学术论坛(2013年2期)2013-05-07 09:51:48
中国烟草学报(2012年4期)2012-04-09 07:11:48
