
脂质沉积性肌病21例临床分析
2014-11-21 06:58西安交通大学医学院第二附属医院神经内科西安710004
陕西医学杂志 2014年7期
西安交通大学医学院第二附属医院神经内科(西安710004)
武 涛# 吴海琴△ 贾 颐# 周金津# 党根喜#
脂质沉积性肌病(Lipid storage myopathy,LSM)是一类因线粒体脂肪酸转运和β-氧化缺陷,致使肌纤维内脂质异常沉积而引起的代谢性肌病,临床相对少见。现将我院收治的21例LSM临床特点、误诊原因、鉴别诊断和治疗报告如下。
临床资料
1 一般资料 选择西安高新医院神经内科从2004年3月至2013年2月期间就诊的21例LSM为研究对象,男性12例,女性9例。发病年龄12~56岁,平均29.33±13.35岁。病程1月至18年不等,平均54.38±4.95月。2例有家族史。14.3%(3/21)病前有感染、腹泻或呕吐等诱因。14.3%(3/21)亚急性起病,85.7%(8/21)慢性起病。
2 临床症状和体征 首发症状:90.5%(19/21)为肢体无力、运动不耐受,9.5%(2/21)为抬头费力。57.7%(12/21)双上肢近端无力,肌力Ⅲ~Ⅳ级;15.4%(3/21)双上肢远端无力,肌力Ⅳ级。66.7%(14/21)双下肢近端无力,肌力Ⅲ~Ⅳ级;9.5%(2/21)双下肢远端无力,肌力Ⅳ级。66.7%(14/21)颈肌无力,42.3%(9/21)咬肌力弱,38.1%(8/21)肌痛,4.8%(1/21)双侧股四头肌萎缩。61.8%(13/21)四肢腱反射减低或消失。9.5%(2/21)双下肢远端麻木感,仅4.8%(1/21)有感觉障碍体征。
3 实验室检查 95.2%(20/21)肌酸激酶(CK)和α-羟基丁酸脱氢酶(HBD)升高。95.2%(20/21)CK值≤2000U/L,4.8%(1/21)CK 值>2000U/L。90.5%(19/21)乳酸脱氢酶(LDH)升高,81.0%(17/21)肌酸激酶同工酶(CK-MB)升高,66.7%(14/21)天门冬氨酸氨基转移酶(AST)升高。治疗后复查,CK恢复最快,LDH和HBD恢复较慢。
4 神经电生理 25.0%(4/16)肌电图为单纯肌源性损害。31.3%(5/16)为肌源性合并神经源性损害。43.7%(7/16)未见异常。100%(11/11)重复频率电刺激检查阴性。
5 肌肉病理 光镜观察(附图)HE染色:15例肌纤维大小均匀,6例肌纤维大小不均匀,少数肌纤维萎缩、变性坏死;21例均在部分肌纤维内见细小筛孔样空泡,少数病例中部分空泡融合成片;均未见炎性细胞浸润。ORO染色:空泡染色阳性。PAS染色糖原成分正常。改良Gomori三色:未见破碎红纤维(RRF)。ATPase染色:空泡样改变累及两型肌纤维,以Ⅰ型肌纤维为主。电镜见肌膜下及肌原纤维间串珠状大小均匀的脂滴沉积。
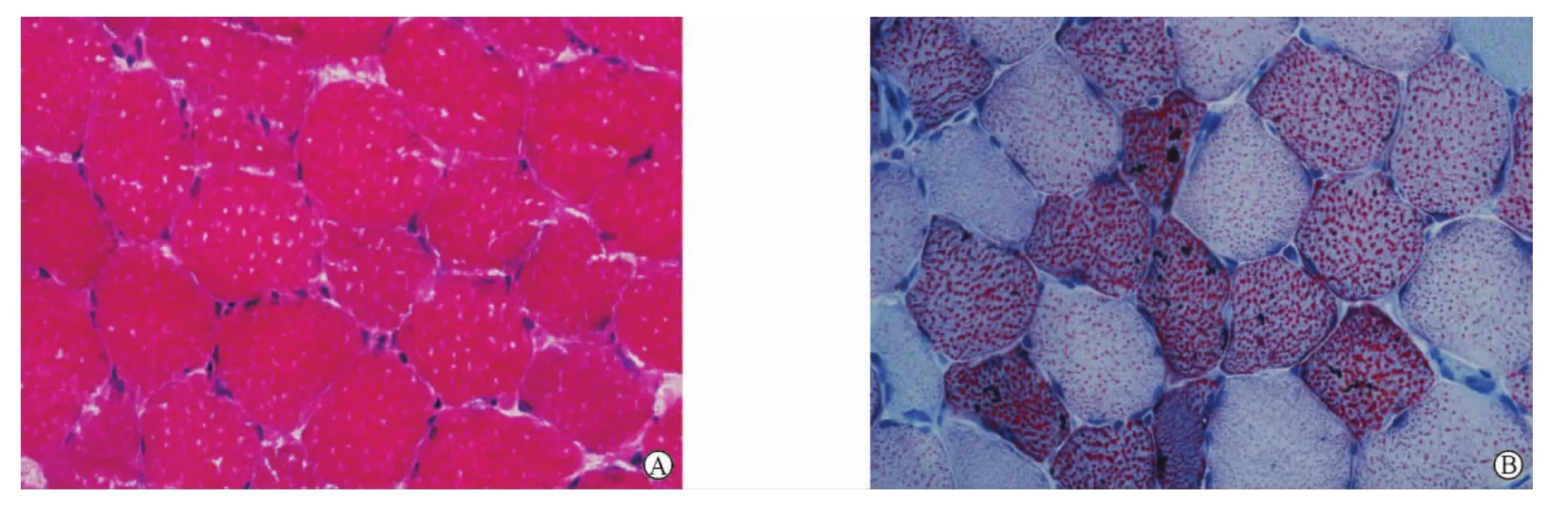
附图 A:部分肌纤维内有空泡样改变(×400,HE染色);B:部分肌纤维内空泡脂滴染色增强(×400,ORO染色)
6 误诊情况 57.1%(12/21)患者曾被误诊,时间从1月至18年,平均41.83±58.51月。4例误诊为多发性肌炎,2例早期误诊为病毒性心肌炎,后误诊为多发性肌炎,2例误诊为病毒性心肌炎,误诊为重症肌无力、慢性炎性脱髓鞘性多发性神经根神经病、进行性肌营养不良、颈椎病者各1例。
7 治疗和预后 全部21例均给予高糖低脂饮食。2例单用核黄素60mg/d口服,9~14d后症状好转。5例核黄素(剂量同前)联合左旋肉碱3g/d口服,5~18d后症状好转。14例地塞米松10~20mg/d静脉注射联合核黄素与左旋肉碱(剂量同前)治疗,7~21d症状好转。
讨 论
脂肪酸在线粒体内进行β-氧化是骨骼肌的重要能量来源。脂肪酸代谢过程中任何环节的缺陷,如甘油三酯的降解、肉碱的摄取、脂肪酸的转运及氧化等均可干扰肌纤维内脂质代谢,导致肌肉能量产生障碍[1],肌纤维内脂质异常堆积。目前已知4种基因诊断的LSM:原发性肉碱缺乏症、多种酰基辅酶A脱氢酶缺陷 病(Multiple acyl-CoA dehydrogenase deficiency,MADD)、中性脂质贮积病伴鱼鳞病、中性脂质贮积病伴肌病[2]。国内LSM多为核黄素反应性 MADD[3],为常染色体隐性遗传病,主要由电子转移黄素蛋白脱氢酶基因缺陷所致。
LSM的临床特征:青少年期或成人发病;慢性或亚急性起病,主要表现为四肢近端无力、运动不耐受,多伴颈肌力弱;症状有波动性,可自发缓解;肌酶轻至中度升高;肌电图呈单纯肌源性损害或合并神经源性损害;多数患者对皮质类固醇激素治疗敏感。
本病四肢近端无力、肌痛、肌萎缩,肌酶升高,肌电图呈肌源性损害,激素治疗敏感等特点,最常被误诊为多发性肌炎(Polymyositis,PM)。和LSM可自发缓解不同,PM临床症状通常不会自发缓解。CK在LSM常<2000U/L[3];而PM 急性期可达正常上限的50倍[4]。PM典型病理特点:肌纤维大小不一,变性、坏死,炎性细胞浸润以CD8+T细胞为主,肌膜表达MHC I分子可确诊[4]。LSM病理特点为:部分肌纤维内脂滴显著沉积,累及两型肌纤维,以Ⅰ型肌纤维受累为主;电镜下肌原纤维间脂滴贮积,部分病例线粒体数量和形态异常[5]。少数LSM见肌纤维坏死与再生,伴细胞吞噬现象[6],当PM继发肉碱缺乏时,少数肌纤维内脂滴增多,给鉴别造成困难。但PM少数肌纤维内继发性脂质增多的程度和肌纤维受累的广泛性,远不及原发性代谢异常,且同时具有PM的典型病理改变可资鉴别。
LSM四肢易疲劳、咀嚼与抬头费力,症状活动后加重而休息后减轻,易误诊为重症肌无力(Myasthenia gravis,MG)。与82.2%MG患者眼外肌受累[7]不同,LSM眼外肌通常不受累[3]。LSM为较持续的运动不耐受,疲劳周期较长;而MG病态疲劳与每天波动性密切相关[7]。MG新斯的明试验阳性率在80%~90%,全身型MG重复频率电刺激阳性率达90%[8],而在LSM则为阴性。鉴别困难者应行肌肉病理检查除外LSM。
LSM患者肌无力可伴感觉异常,合并神经源性损害时运动单位动作电位时限增宽、波幅增加,运动及感觉神经传导速度减慢,感觉神经动作电位波幅降低[5,6]。上述特点使其易误诊为慢性炎性脱髓鞘性多发性神经根神经病,但该病经典型的症状限于周围神经,感觉障碍突出;电生理检查神经传导速度减慢或异常波形离散;脑脊液蛋白-细胞分离;腓肠神经活检节段性脱髓鞘,轴索变性等表现;病理见肌纤维群组化萎缩、变性与坏死,ORO染色肌浆内无脂滴或仅部分增多[9]。
肢带型肌营养不良与LSM有共同点:四肢近端肌无力、肌酶升高、肌电图呈肌源性损害。但前者肌无力呈缓慢进行性加重,激素治疗无效;病理见肌纤维大小不等、坏死及再生,肌纤维分裂、漩涡状纤维等。与LSM病理发现脂滴沉积、油红反应增强可鉴别。
LSM因活动后疲乏、胸闷气短、肌酶升高,心电图示窦性心动过速等,常误诊为病毒性心肌炎。LSM心悸胸闷可能为肉碱缺乏致长链脂肪酸堆积而供能障碍,影响心肌收缩功能所致[9]。
因此,LSM临床表现缺乏特异性,临床极易误诊[10]。诊断有赖于肌肉病理检查。鉴于国内以核黄素反应性MADD为主,可首选核黄素治疗[3],效果不佳者补充左旋肉碱、辅酶Q10或皮质类固醇激素治疗。注意避免劳累、上呼吸道感染、禁食等诱因。
[1]Laforet P,Vianey-Saban C.Disorders of muscle lipid me-tabolism:diagnostic and therapeutic challenges[J].Neuromuscul Disord,2010,20(11):693-700.
[2]Liang WC,Nishino I.Lipid storage myopathy[J].Curr Neurol Neurosci Rep,2011,11(1):97-103.
[3]焉传祝,卢家红.我国脂质沉积性肌病的病因研究历程[J].中华神经科杂志,2011,44(5):300-303.
[4]中华医学会风湿病学分会.多发性肌炎和皮肌炎诊断及治疗指南[J].中华风湿病学杂志,2010,14(12):828-831.
[5]陈昆明,李剑敏,方周溪.脂质沉积性肌病的临床和病理特点分析[J].中国实用神经疾病杂志,2008,11(7):87-88.
[6]陈 琳,郭玉璞,任海涛,等.貌似多发性肌炎的脂质沉积性肌病病理改变[J].中华神经科杂志,2001,34(2):81-83.
[7]丛志强,李海峰.重视重症肌无力的临床诊断[J].中华神经科杂志,2006,39(11):786-788.
[8]崔丽英.应重视重症肌无力的临床诊断和规范化治疗[J].中华神经科杂志,2007,40(8):505-506.
[9]梅海云,杨晓苏,肖 波,等.脂质沉积性肌病诊断及误诊分析[J].卒中与神经疾病,2009,16(3):168-171.
[10]高丽霞,陈翠英.脂质沉积性肌病1例[J].陕西医学杂志,2006,35(11):4568.
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26
中国临床医学影像杂志(2022年5期)2022-07-26
国际放射医学核医学杂志(2021年10期)2021-02-28
世界科学技术-中医药现代化(2020年2期)2020-07-25
国际放射医学核医学杂志(2020年2期)2020-05-30
中国中医急症(2019年10期)2019-05-21
中国畜牧杂志(2019年4期)2019-04-20
家庭百事通·健康一点通(2017年3期)2017-03-22
现代检验医学杂志(2016年4期)2016-11-15
动物营养学报(2015年9期)2016-01-07
