
电解锰废水中Cr 6+、Mn 2+的去除方法研究
2014-11-16 02:16张慧卿周康根吴运东
有色金属科学与工程 2014年3期
张慧卿, 周康根, 吴运东
(1.中南大学冶金与环境学院环境工程研究所,长沙410083;2.国家重金属污染防治工程技术研究中心,长沙410083)
电解锰工业是一项资源、能源消耗高,污染物产生量大的工业行业,在其生产过程中,需要排放大量的工业废水,每生产1 t电解锰,大约需要排放工业废水350 t[1].我国现有电解锰企业126家,电解锰产量已超过100万t/a,产生的废水约3.25亿t/a[2].电解锰废水水质复杂,含有高浓度Cr6+、Mn2+等有害成分,悬浮物多,色度大,若不进行合适的处理将对环境造成严重的污染[3].电解锰废水的主要成分及浓度如表1所示[4].

表1 电解锰废水的主要成分及浓度
从表1可以看出,电解锰废水中的主要污染物Mn2+超标 750~1 250 倍,Cr6+超标 300~700 倍,NH3~N超标50~100倍.
目前,已经在生产中应用的电解锰废水处理技术主要有还原中和沉淀吹脱法[5]、离子交换法[6]和全过程控制技术[7-8].还原中和沉淀吹脱法处理成本高,有二次污染,Mn2+、Cr6+难以回收利用[9];离子交换法应用于电解锰废水领域,对树脂的强度、耐用性和连续使用周期有着较高要求[3];全过程控制技术中,采用焦亚硫酸钠作为还原剂,其溶液暴露在空气中易被氧化,故实际操作中,焦亚硫酸钠溶液要现配现用,固体焦亚硫酸钠也要妥善存放,以免失效[8].因此,开发经济高效的新型电解锰废水处理技术,已成为电解锰行业可持续发展的当务之急.
还原沉淀法除Cr6+具有一次性投资少,运行费用低,处理效果好,操作管理简便等优点[10],因而在工业上得到广泛的应用.常用的还原剂有SO2,FeSO4,Fe,NaH2SO3,Na2SO3等.其中,SO2还原法需增加硫磺燃烧炉制造SO2气体,且硫磺消耗量很大,设备易腐蚀,需加强防毒设施[11];FeSO4还原法需耗用大量的FeSO4、苛性钠及蒸汽等,运行费用高[12];铁屑还原法出水水质发黄,处理设施较大,污泥量多,不易处理[13].NaH2SO3还原法实验药剂供应不普遍,处理费用高[14];Na2SO3还原法是当前含铬废水处理工艺中最为环保节能的方法之一,既可保证出水水质达标,又能够回收 Cr(OH)3,设备和操作简单,节省经费,节约占地面积[15].由于Cr6+在酸性和碱性废水中都以稳定的离子存在,不能用简单的氢氧化物沉淀法除去,因此本研究先用Na2SO3将Cr6+还原为Cr3+,再用碱中和使Cr3+生成Cr(OH)3沉淀,从而使铬从废水中去除.
曝气氧化技术广泛应用于生活污水和工业废水的好氧生物处理中[16].通过曝气除锰技术处理电解锰废水已有报道,谌永红等采用还原中和-混凝沉淀-曝气吹脱工艺处理电解金属锰含铬废水及尾矿库含锰渗滤液,出水 Cr6+、Mn2+能够达到排放标准,但对废水中 Cr6+、Mn2+的去除率仅为 81%和72%[17].针对曝气除锰不能达到100%的Mn2+去除率和锰不能实现资源回用的弊端,本研究提出一种新思路,投加少量的MnO2做晶种曝气除锰,晶种曝气除锰的效率远比单纯的曝气除锰效率高.在合适的条件下,通过将Mn2+转化为MnO2沉淀,在除去废水中锰的同时,生成的MnO2可继续作为催化剂和吸附剂在反应器中循环使用,更有效地回收利用了锰.
基于以上分析,研究开发了一种新型的用于电解锰废水治理的组合新工艺:Na2SO3还原法除铬-MnO2做晶种曝气除锰.即先采用Na2SO3做还原剂还原Cr6+后再调节pH值除铬-MnO2做晶种曝气除锰组合新工艺处理模拟电解锰废水,并且验证了最佳工艺条件,为电解锰废水的处理提供了一种新的经济有效的方法.
1 实验部分
1.1 实验试剂
重铬酸钾、浓盐酸、氢氧化钠、硫酸铵、硫酸锰,均为分析纯.
1.2 实验仪器及装置
实验仪器主要有:320型 pH计(METTLER TOLEDO),1200RT 搅拌器(Type Heidon),TAS 型原子吸收分光光度计(北京普析通用公司),PM2500型电子天平 (日本シイベルヘグナー株式会社),DZF-6050型真空干燥箱 (上海精宏实验设备有限公司),RAT-1水浴恒温反应釜 (上海顺义生物仪器有限公司),SHZ-82A水浴恒温振荡器 (金坛市医疗仪器厂);实验装置见图1.
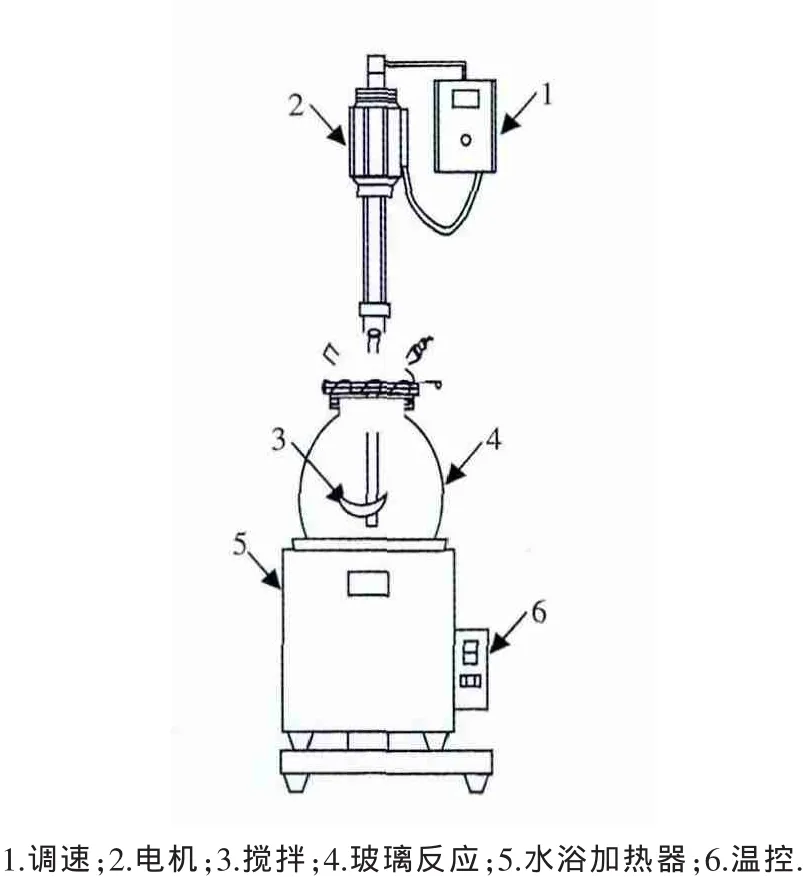
图1 Na2SO3做还原剂除铬实验装置
1.3 实验废水
模拟废水:Cr6+100 mg/L,Mn2+1 000 mg/L,NH4+1 000 mg/L.
实际废水:实验用水取自湖南某电解锰企业生产过程中排放的电解锰废水,分析结果如表2所示.

表2 实际电解锰废水分析结果
1.4 实验方法
实验于25℃恒温水浴锅中进行.取1 L模拟电解锰废水倒入反应釜中,投加一定量的Na2SO3后调节pH值,按转速250 r/min搅拌一定时间后,调节pH值至碱性使Cr(OH)3沉淀完全.静置20 min后过滤,取上清液测定铬锰浓度.剩余上清液加入MnO2做晶种,调节pH至碱性,在50℃水浴中曝气,控制空气流量为450 L/h,过滤后,取上清液测定铬锰浓度.
1.5 分析方法
pH值采用玻璃电极法测定;总铬、Mn2+浓度采用火焰原子吸收分光光度法测定[18-19];Cr6+浓度采用二苯碳酰二肼分光光度法测定[20].
重金属 B(%)去除率由式(1)[21]计算:
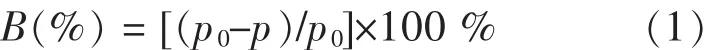
式(1)中:p0为反应前,水样中重金属的质量浓度,mg/L;p为反应后,水样中重金属的质量浓度,mg/L.
2 结果与讨论
2.1 Na2SO 3用量对Cr 6+转化率的影响
Na2SO3作为还原剂将 Cr6+还原为 Cr3+,Cr3+在碱性条件下以Cr(OH)3沉淀形式去除.当废水中Cr6+浓度在50~100 mg/L时,Na2SO3添加量是废水中Cr6+含量的7~11倍时才能还原完全;且废水中Cr6+浓度愈高,Na2SO3添加量愈小[8],这是废水中含有溶解氧和其他氧化性杂质所造成的.据此,本研究所配制模拟废水中的Cr6+浓度为100 mg/L,理论需投加1.1 g Na2SO3, 故本实验取 Na2SO30 g,0.5 g,1g,1.5 g,2 g分别进行实验,结果如图2所示.
由图2可知,当Na2SO3用量为0.5 g/L时,Cr6+转化率已达99.76%,溶液中剩余Cr6+浓度为0.24 mg/L,考虑到后续MnO2做晶种曝气除锰环节,MnO2作为吸附剂仍可吸附少部分第一步未除尽的Cr6+,故此处Na2SO3的最佳投加量为0.5 g/L.由此看出,处理模拟电解锰废水的Na2SO3实际投加量远小于理论投加量,这是由于理论投加量是只有Cr6+存在的含铬废水计算而得的,而本研究是多离子体系的电解锰废水,离子间的相互作用势必对实验结果有影响,有待于进一步考证.
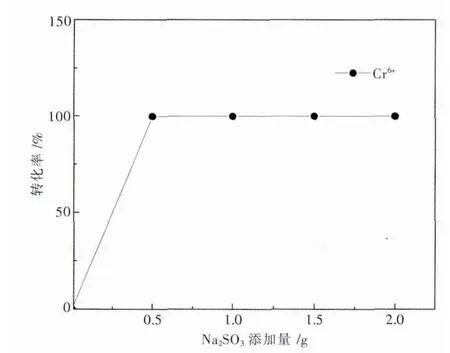
图2 Na2SO3用量对Cr 6+转化率的影响
2.2 还原反应时间对Cr 6+转化率的影响
当pH值在2.2~3.0时,还原反应在5 min已基本趋于平衡,还原率达到99.88%,此后随还原反应时间的延长,Cr6+含量呈缓慢变化趋势[9].故本实验分别于还原 反 应 时 间 为 1 min,2 min,4 min,6 min,8 min,10 min取样测Cr6+浓度,并计算Cr6+转化率,实验结果如图3所示.
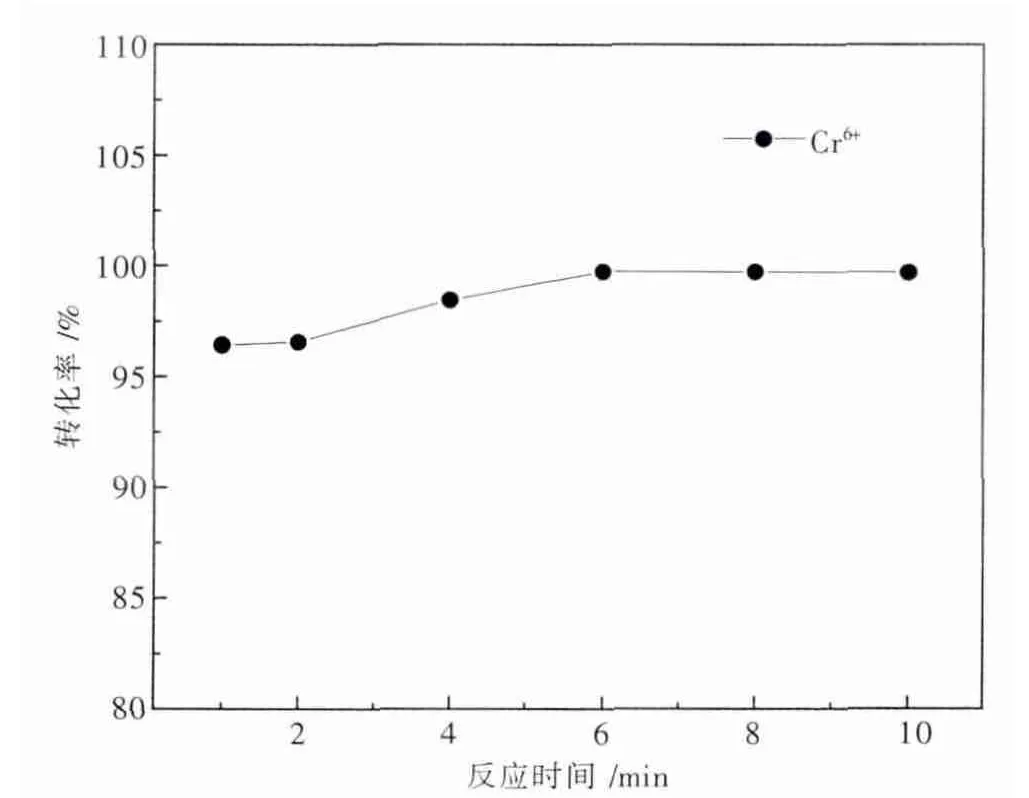
图3 还原反应时间对Cr 6+转化率的影响
由图3可知,随还原反应时间的延长,Cr6+转化率逐渐升高,直至还原反应时间达6 min时,Cr6+转化率为99.74%,并趋于稳定.说明还原反应进行6 min,溶液中的Cr6+已基本转化为Cr3+,再增加还原反应时间对于Cr6+转化率并无多大影响,故最佳还原反应时间为6 min.
2.3 还原反应p H值对Cr 6+转化率的影响
还原反应在酸性条件下进行,故本实验取还原反应 pH 值为 2,3,4,5,6分别进行实验,结果如图 4所示.
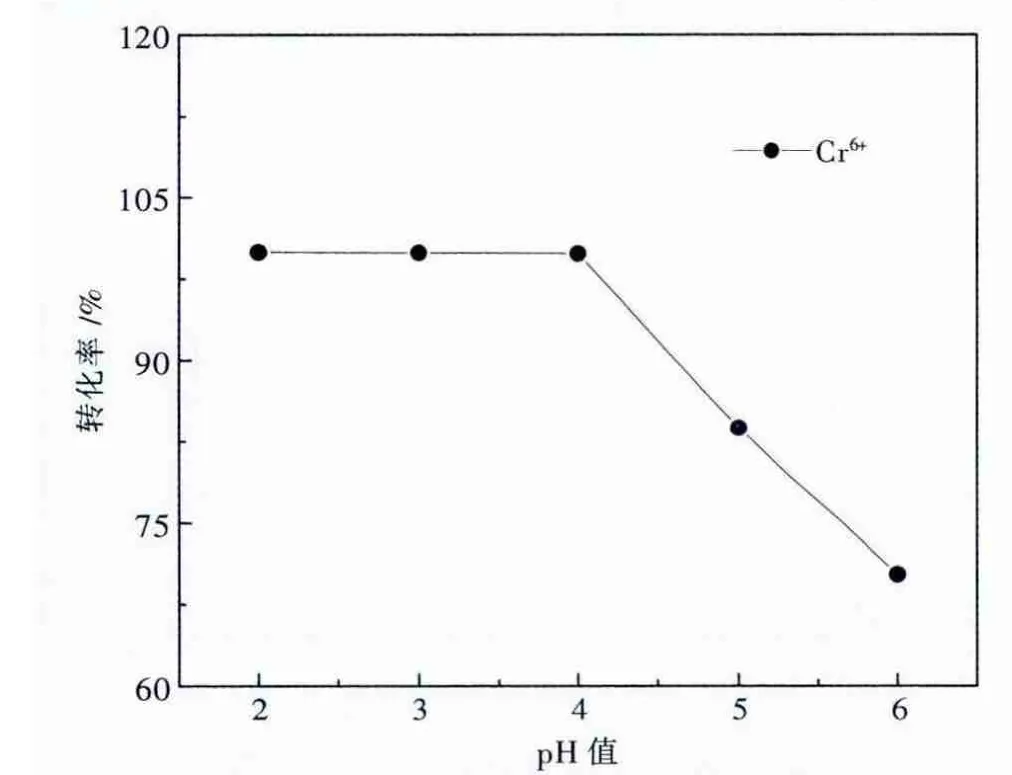
图4 还原反应p H值对Cr 6+转化率的影响
由图4可知,随还原反应pH值的增大,Cr6+转化率逐渐降低,特别当还原反应pH值大于4后,Cr6+转化率急剧下降.Na2SO3还原Cr6+反应pH值不宜大于4,否则反应速度很慢[8].这是由于随着Na2SO3的不断投入,废水的pH值会逐渐提高,使得Cr6+的还原速度减慢,即使投加过量Na2SO3,还原反应仍不能进行彻底,这时加碱沉淀后的出水会发黄.同时,随着废水的 pH 值升高,Cr(OH)3会过早产生,形成胶体,当下一步加碱时,Cr(OH)3不能很快沉淀,需要重新用酸将沉淀溶解,然后再加碱才能使Cr(OH)3顺利沉淀.故最佳还原反应pH值为4.
2.4 污泥沉淀时间对出水总铬浓度的影响
Cr6+转化为 Cr3+后,在碱性条件下将生成 Cr(OH)3沉淀去除,此处污泥的主要成分即为Cr(OH)3.本实验分别于沉淀时间为 0 min,2 min,5 min,8 min,10 min,15 min,25 min取样测出水总铬浓度,实验结果如图5所示.
由图5可知,污泥沉淀时间为8 min时,出水总铬浓度仅为0.062 mg/L,当沉淀时间大于10 min时,出水总铬浓度低于检测限.由于本电解锰废水体系中有NH3-NH4+的缓冲体系存在,使投加酸碱调节pH值时,废水体系的pH值变化速度较慢,故取污泥最佳沉淀时间为10 min.
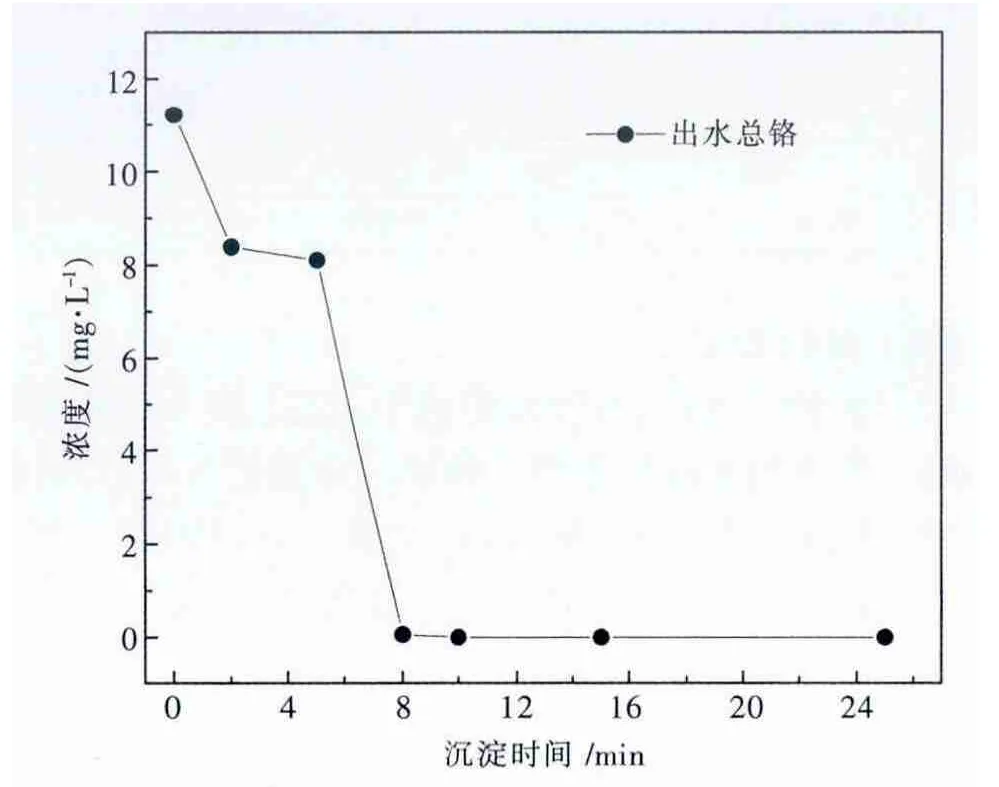
图5 污泥沉淀时间对出水总铬浓度的影响
2.5 污泥沉淀p H值对出水总铬浓度的影响
Cr6+还原后所生成的Cr3+在碱性条件下生成沉淀,主要成分为Cr(OH)3,故本实验分别取沉淀反应pH 值为 6,7,8,9,10 进行实验,结果如图 6 所示.

图6 污泥沉淀p H值对出水总铬浓度和污泥体积的影响
由图6可看出,污泥沉淀pH值为8时,出水总铬浓度仅为0.012 mg/L,污泥体积仅为150 mL.当污泥沉淀pH值大于8时,出水总铬浓度低于0.012 mg/L,但污泥体积远大于150 mL,说明污泥沉淀pH值越高,出水总铬浓度越低,但污泥体积会逐渐增大.这是由于Cr(OH)3沉淀过程中溶液pH值会上升,易形成胶体,导致污泥不能快速沉淀.另外,若体系pH值太高,Cr(OH)3沉淀后的溶液在下一步的曝气实验中还需要另外调节pH值,所以最佳污泥沉淀pH值为8.
2.6 晶种MnO 2用量对出水重金属离子浓度的影响
本实验取除铬后的模拟废水分别加入晶种MnO210 g,15 g,20 g,25 g,30 g 进行曝气除锰实验,结果如图7所示.
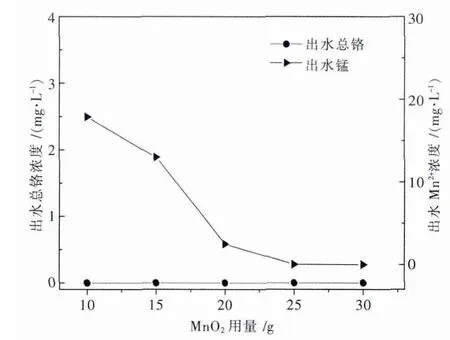
图7 MnO2用量对出水重金属离子浓度的影响
由图7可知,MnO2用量大于10 g时,出水总铬浓度低于检测限,其原因可能为Na2SO3还原中和沉淀除铬彻底,或者是新生成的MnO2具有一定的吸附能力,将第一步未除尽的少量铬吸附除去.出水Mn2+浓度也随着MnO2用量的增加而逐渐降低,当MnO2用量高于25 g时,出水锰浓度低于0.046 mg/L.
实验中发现,曝气后澄清的出水放置一段时间后会有黑褐色细小颗粒生成,说明溶液中的Mn2+会被空气氧化而溶出,造成出水浑浊,且黏附在容器内壁的锰氧化物也很难去除,所以为了避免出水Mn2+超标且出水水质清晰,晶种曝气应彻底去除溶液中的Mn2+,所以此处选择MnO2最佳投加量为25 g.
2.7 晶种曝气p H值对出水Mn2+浓度的影响
配制2组各5瓶1 L的Mn2+浓度为1 000 mg/L的电解锰废水,一组加入25 g的MnO2做晶种,一组不添加 MnO2, 分别调节曝气 pH 值为 8、9、10、11、12,置于50℃水浴锅中,控制空气流量为450 L/h,曝气反应10 min,取出一定量的水样,过滤后分析水样中Mn2+浓度,实验结果如图8所示.
由图8可知,添加MnO2做晶种,晶种曝气pH值为9时,出水Mn2+浓度仅为1.07 mg/L,当晶种曝气pH值大于9时,出水Mn2+浓度低于1.07 mg/L;而不添加晶种的处理组在曝气pH值达到12以后出水Mn2+浓度才低于1 mg/L.在pH值相同的情况下,添加MnO2做晶种比不添加晶种曝气反应的出水Mn2+浓度要低.但无论是否添加MnO2做晶种,增大pH值均可提高Mn2+去除率.在pH=9时,添加晶种处理组的出水Mn2+浓度即达到了1.07 mg/L,继续提高pH,出水Mn2+浓度变化不大,故最佳晶种曝气pH值为9.
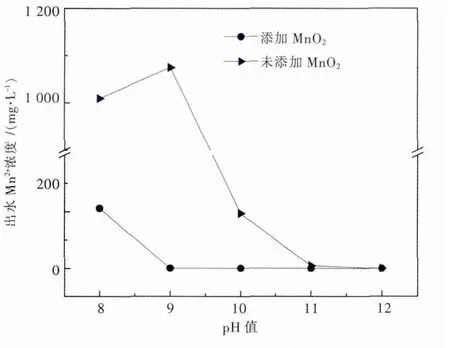
图8 添加MnO2与不添加MnO2做晶种体系曝气p H值对出水Mn2+浓度的影响
2.8 晶种曝气时间对出水Mn2+浓度的影响
由图9可看出,不添加MnO2做晶种曝气的处理组,出水Mn2+浓度远高于添加MnO2的处理组,说明添加MnO2做晶种可以高效去除溶液中的Mn2+.
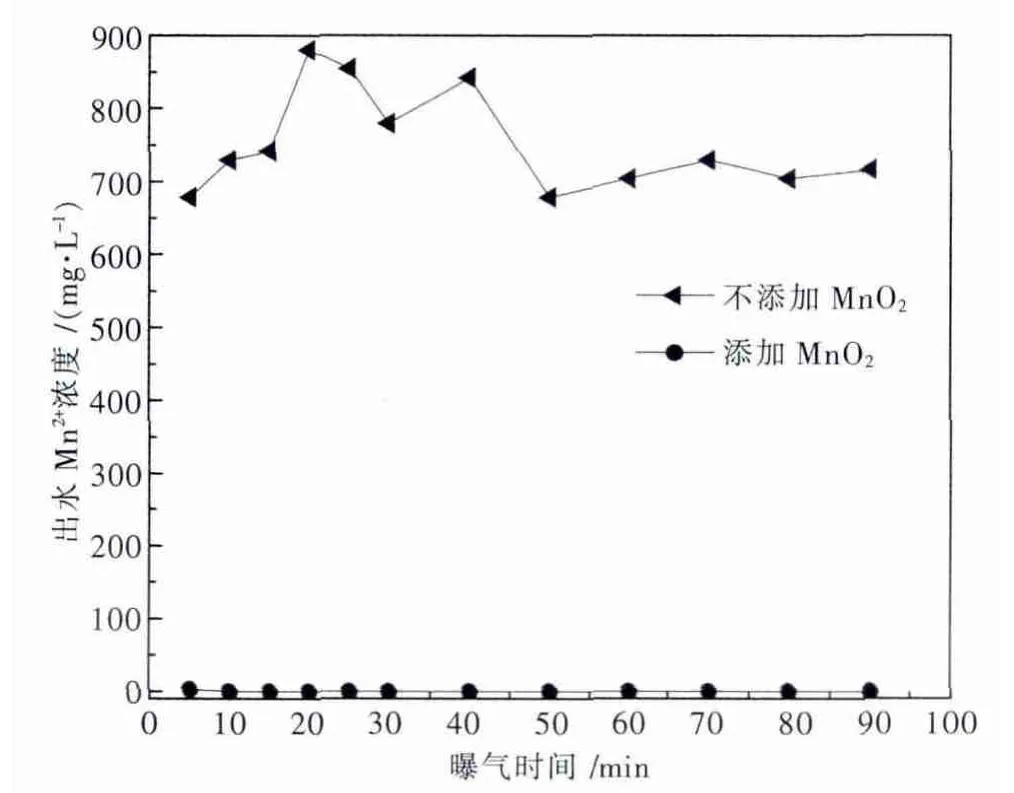
图9 添加MnO2与不添加MnO2做晶种体系曝气时间对出水Mn2+浓度的影响
实验中发现,加入MnO2后调完pH值,出水Mn2+浓度已经降到7 mg/L,继续曝气10 min出水Mn2+浓度降至0.02 mg/L,当曝气时间大于10 min时,出水Mn2+浓度低于检测限.推断是因为加入的MnO2吸附作用使得溶液中的Mn2+浓度快速降低.MnO2沉淀物的等电点是pHz=2.8±0.3,实验中溶液pH高于等电点,MnO2表面发生酸性离解,而致表面电荷为负.氧化物呈负电荷表面时,能对水中阳离子进行离子交换吸附,所以MnO2能吸附水中Mn2+,并且吸附速度很快.而且反应生成物MnO2对Mn2+的氧化反应有催化作用,就使得吸附在MnO2表面的Mn2+又被氧化成MnO2,继续做催化剂使用.由于在曝气10 min后溶液中的Mn2+浓度已基本稳定并低于检测限,故本实验取最佳曝气时间为10 min.
2.9 化学沉淀-晶种曝气组合工艺处理电解锰废水的最佳工艺条件实验
据以上单因素条件实验结果,分别对实验室模拟电解锰废水和实际电解锰废水进行还原中和沉淀-晶种曝气组合工艺的最佳工艺条件实验:Na2SO3加入量0.5 g/L,调pH值至4后进行还原反应6 min;将pH值调至8进行沉淀反应10 min.静置20 min,过滤,取上清液调pH值至9,加入25 g/L MnO2做晶种,50℃水浴曝气10 min,控制空气流量为450 L/h,过滤,取上清液测定铬锰浓度,实验结果如表3所示.
由表3可看出,采用化学沉淀-晶种曝气组合工艺处理实验室模拟电解锰废水实验中,还原反应阶段Cr6+转化率高达99.93%,污泥体积仅为180 mL,利于后续压缩处置,废水经处理后,最终出水总铬浓度从100 mg/L降至0.023 mg/L,Mn2+浓度从1 000 mg/L降至0.033 mg/L,总铬去除率高达99.98%,Mn2+去除率高达100%,均达到我国 《污水综合排放标准(GB8978-1996)》一级标准.

表3 最佳工艺条件实验结果
而对于采自湖南某电解锰企业生产过程中排放的实际电解锰废水实验中,最终出水总铬浓度从100 mg/L降至0.5 mg/L,Mn2+浓度从1 000 mg/L降至0.4 mg/L,总铬去除率高达99.5%,Mn2+去除率高达99.96%,由此可见,用化学沉淀-晶种曝气组合工艺处理实际电解锰废水,可取得较好的处理效果,处理后的废水可直接达标排放.
故化学沉淀-晶种曝气组合工艺去除电解锰废水中Cr6+和Mn2+的最佳工艺条件为:Na2SO3投加量0.5 g/L,还原反应pH值为4,还原反应时间6 min,污泥沉淀pH值为8,污泥沉淀反应时间10 min,晶种MnO2投加量25 g/L,晶种曝气pH值为9,晶种曝气时间10 min.
3 结 论
1)本研究采用化学沉淀-晶种曝气组合新工艺处理电解锰废水分3个阶段:还原反应阶段,Na2SO3投加量0.5 g/L,还原反应pH值为4,还原反应时间6 min;污泥沉淀反应阶段,污泥沉淀pH值为8,污泥沉淀反应时间10 min;晶种曝气阶段,25 g/L的MnO2做晶种,晶种曝气pH值为9,在50℃水浴中曝气10 min,控制空气流量450 L/h.
2)电解锰废水采用常规处理方法,出水重金属难达标,且不能实现资源的有效回用,本研究晶种曝气除锰环节,投加的MnO2做晶种,催化氧化溶液中Mn2+快速生成MnO2,可继续回用做催化剂和吸附剂,在工业应用的连续实验中,既节省了原料成本,又减少了尾端处理的繁琐工艺.
3)本研究先除铬后除锰的设计思路,充分考虑到MnO2具有一定的吸附功能,若是第一步除铬不彻底,遗留了一部分重金属铬,那么第二步MnO2做晶种曝气除锰环节,重金属铬锰可一并彻底去除.
4)化学沉淀-晶种曝气组合新工艺所用设备简单,投资少,沉渣可综合利用,适于工业化应用.
[1]谭柱中,梅光贵,李维健,等.锰冶金学[M].长沙:中南大学出版社,2004,322-324.
[2]杜兵,周长波.电解锰废水处理技术现状及展望[J].工业水处理,2010,30 (12): 34-36.
[3]徐婷婷,杨敏,于旭青,等.离子交换技术在电解锰工业废水处理中的应用[J].污染防治技术,2009,22(3):62-64.
[4]魏建.电解锰废水的处理及资源化利用研究[D].北京:中国矿业大学,2009.
[5]Tu Y J,Chang C K,Wang S L,et al.Treatment of complex heavy metal wastewater using a multi-staged ferrite process[J].Journal of Hazardous Materials,2012,50(1): 379-384.
[6]付杰,李燕虎,叶长森,等.DMF在大孔吸附树脂上的吸附热力学及动力学研究[J].环境科学学报,2012,32(3):639-644.
[7]陆文忠,许灵群,李国平.电镀废水处理自动控制技术的应用[J].能源环境保护,2003,17(2):38-40.
[8]马晓鸥,康思琦,尹庚明,等.电镀废水处理装置工艺条件的优化[J].五邑大学学报(自然科学版),2004,18 (1): 28-31.
[9]汪启年,王璠,高小娟.我国电解金属锰废水处理技术研究进展[J].中国锰业,2011,29 (2):10-14.
[10]熊道陵,李英,钟洪鸣,等.铬回收技术及其研究进展[J].有色金属科学与工程,2011,2(5):6-11.
[11]许闻旎.电镀含铬废水处理技术探讨[J].鞍钢技术,2001,1(6):59-62.
[12]胡晓峰.二氧化硫法处理含铬废水[J].江西化工,2009,1(2):125-127.
[13]谢少雄,黄功浩,黄美燕.铁屑法处理电镀含铬废水的试验及应用[J].工业水处理,2003,23(6):28-30.
[14]刘存海,朱玉凤,张光华.含铬废水处理技术概况及进展[J].辽宁化工,2009,38 (11): 811-813.
[15]丛皓,赵永权.电镀废水处理的新工艺与流程[J].节能,2009,28(2): 9-10.
[16]崔福义,张兵,唐利.曝气生物滤池技术研究与应用进展[J].环境污染治理技术与设备,2005,6(10):1-7.
[17]谌永红,李伟明,刘航.含铬、锰酸性废水处理技术探讨[J].环境与可持续发展,2004,1(1):18-20.
[18]叶军.火焰原子吸收分光光度法测定废水中总铬[J].环境工程,2005,23 (1): 61-62.
[19]国家环境保护总局.水质 锰的测定 甲醛肟分光光度法 (试行)[M].北京:中国环境科学出版社,2007.
[20]国家环境保护总局.水和废水监测分析方法[M].4版.北京:中国环境科学出版社,2002.
[21]鲁秀国,刘秀兰,梁淑轩,等.TiO2掺杂Pb2+吸附剂去除废水中Cr6+的实验研究[J].工业用水与废水,2002,33(5): 27-29.
猜你喜欢
化学工业与工程(2022年4期)2022-09-20
石油炼制与化工(2022年9期)2022-09-05
四川化工(2021年6期)2022-01-12
节能与环保(2021年9期)2021-10-19
冶金经济与管理(2020年4期)2020-09-28
昆钢科技(2020年6期)2020-03-29
山东化工(2020年16期)2020-02-20
应用化工(2019年10期)2019-11-05
资源节约与环保(2018年1期)2018-02-08
环境保护与循环经济(2017年6期)2018-01-22
