
氮掺杂还原氧化石墨烯负载铂催化剂的制备及甲醇电氧化性能
2014-09-21 08:59马俊红
物理化学学报 2014年7期
王 丽 马俊红,2,*
(1新疆大学化学化工学院,乌鲁木齐830046;2新疆大学应用化学研究所,先进功能材料自治区级重点实验室,乌鲁木齐830046)
1 引言
直接甲醇燃料电池(DMFC)因具有能量转化效率高、环境友好等特点,被视为21世纪最有潜力的新型能源.然而,目前DMFC使用的Pt基催化剂的成本高和稳定性较差限制了DMFC的商业化应用.当前DMFC主要使用的是碳承载Pt系列催化剂(Pt/C),该类催化剂使用高比表面积的碳载体材料实现催化剂活性组分Pt纳米颗粒的良好分散,与此同时,通过改变碳载体的表面结构和石墨化程度还能间接调变Pt电极催化剂的稳定性.目前常用的碳载体材料主要有碳黑、碳微球、碳纤维、碳纳米管,以及近些年来发现的石墨烯等.而石墨烯因具有独特的石墨化平面结构、高比表面积和良好的导电性等众多优异性质,有望成为DMFC电极催化剂的理想载体.
近来的研究表明,使用不同的杂原子,如N、P、B、S等对石墨烯进行掺杂,能够明显影响石墨烯的物理、化学及电子性质,1,2其中,对石墨烯进行N掺杂,一方面可以实现对石墨烯碳材料表面的电荷分布及表面缺陷程度的有效调变,3另一方面掺杂的N还对石墨烯负载的Pt催化剂纳米粒子的成核和生长,4,5以及Pt―C之间的相互作用产生影响,6从而进一步改变石墨烯基Pt电催化剂的性能.因此,N掺杂石墨烯碳材料作为电极催化剂的载体显示出很大的应用潜力.7-9目前实现石墨烯N掺杂的方法很多,如化学气相沉积(CVD)法,10该法能够通过精密的实验参数的控制实现以吡啶型氮和石墨型氮为主的N掺杂石墨烯的制备,但该种方法的产量较低.再如N2等离子处理法、11电弧放电法、12水热法13等.这些方法很难控制N掺杂石墨烯中氮的键合构型,同时还存在制备过程复杂、操作成本高或使用有毒前驱体等不利因素.高温热解法是一种在含氮前驱体存在的情况下高温热解氧化石墨烯制备N掺杂的还原氧化石墨烯碳材料(N-RGO)的方法,该法由于氮源种类丰富(可通过改变氮源的类型实现掺杂氮的键合构型的调变)、14,15操作简单、可进行大规模生产并且条件可控等优点目前被广泛应用.16,17聚苯胺结构中含有大量吡啶型氮,同时在结构上与石墨烯具有相似性,高温热解聚苯胺-氧化石墨烯复合物有利于形成以石墨氮为主的N掺杂的石墨烯纳米层结构,该类N掺杂石墨烯碳材料目前主要是直接作为氧还原反应的电催化剂18或锂离子电池材料,19以其作为电极催化剂载体的研究工作还鲜有报道.本文首先将苯胺原位化学聚合在氧化石墨烯(GO)表面,形成聚苯胺修饰的氧化石墨烯(PANI-GO),然后通过将PANI-GO高温热解实现了GO的热分解还原及其N掺杂,得到了N掺杂的还原氧化石墨烯碳材料(N-RGO),最后以N-RGO负载Pt制备了Pt/NRGO电极催化剂,考察了该电极催化剂对甲醇电氧化反应的催化性能.
2 实验部分
2.1 原料与试剂
石墨粉(上海市阿拉丁化学有限公司,分析纯),氯铂酸(上海精细化工材料研究所,分析纯),硼氢化钠(天津市福晨化学试剂厂,分析纯),过硫酸铵(天津市百世化工有限公司,分析纯),苯胺(天津市致远化学试剂有限公司,分析纯),硫酸(天津市盛淼精细化工有限公司,分析纯),甲醇(西安化学试剂厂,分析纯).Nafion乙醇溶液,5%(w)(安徽瑞邦新能源科技有限公司),双氧水(质量分数30%)(天津市天力化学试剂厂,分析纯),高锰酸钾(上海市化学试剂厂,分析纯),五氧化二磷(天津欧博凯化工有限公司,分析纯),实验中均采用二次去离子水.
2.2 GO的制备
采用改进的Hummer法20制备氧化石墨烯(GO):2.5 g(NH4)2S2O8和2.5 g P2O5加入到12 mL H2SO4(98%)中,将温度升至80°C使固体溶解,再加入3 g石墨粉,在80°C下继续搅拌6 h后冷却至室温,然后加入500 mL水放置一晚;过滤洗涤以上得到的样品,在空气中干燥6 h后得到预氧化的石墨.在冰浴条件下在预氧化石墨中加入120 mL浓硫酸,并缓慢加入15 g KMnO4,然后将温度升至35°C下搅拌2 h后,在室温下继续搅拌5 d,然后加入30 mL 30%H2O2,放置一晚.依次用5%HCl和去离子水洗涤至滤液呈中性,将固体样品在-45°C冷冻干燥48 h,得到GO.
2.3N-RGO的制备
称取GO 0.2 g置于50 mL 0.5 mol·L-1盐酸中,超声波分散处理1 h,得悬浮液.将悬浮液置于冰浴中,滴入250 μL苯胺,搅拌15 min后缓慢加入1.5 mL的1.5 mol·L-1(NH4)2S2O8,在冰浴下继续搅拌6 h,过滤,洗涤,90°C真空干燥3 h得到PANI-GO,将PANI-GO在N2氛围中800°C焙烧1 h得到N-RGO.GO在相同条件下高温处理得到热分解还原的氧化石墨烯碳材料(RGO).
2.4Pt/N-RGO及Pt/RGO的制备
称取50 mg N-RGO于50 mL的蒸馏水中,超声波分散处理1 h.随后在搅拌下逐滴加入1.5 mL 10 mg·mL-1的H2PtCl6·6H2O乙二醇溶液,再缓慢加入新配置的NaBH4溶液,室温搅拌24 h后过滤,洗涤,干燥6 h得到Pt/N-RGO催化剂.Pt/RGO样品的制备方法同Pt/N-RGO,只是将N-RGO替换为RGO.
2.5 材料表征与性能测试
X射线粉末衍射(XRD)测试使用德国Bruker公司D8 Advance型X射线衍射仪器,Cu Kα辐射(λ=0.15418 nm),管电压为40 kV,管电流为100 mA.样品表面形貌分析使用日本日立公司H-600透射电子显微镜(TEM),加速电压为100 kV.拉曼光谱分析采用德国Bruker公司SENTERRA拉曼仪.X射线光电子能谱(XPS)在分析PEPE PHI5300X光电子能谱仪进行测试,使用Mg Kα靶(1253.6 eV),功率为250 W(12.5 kV×20 mA),本底真空度优于10-7Pa,数据处理采用XPSpeak4.1软件进行曲线拟合.催化剂中金属载量使用美国Thermo Fisher公司IRIS Intrepid II XSP型电感耦合等离子发射光谱仪(ICP-AES)分析确定.
电化学测试所用仪器为CHI660B电化学工作站(上海辰华公司),采用三电极体系,以涂覆有催化剂的玻碳电极为工作电极,饱和甘汞(SCE)电极为参比电极,铂丝电极(ϕ=0.5 mm)为辅助电极.甲醇电氧化反应的循环伏安及计时电流测试(0.40 V,vs SCE)在 0.5 mol·L-1CH3OH+0.5 mol·L-1H2SO4电解质溶液中进行.一氧化碳溶出反应在0.5 mol·L-1H2SO4电解质溶液中进行.文中所述电势均为相对于饱和甘汞电极而言.
3 结果与讨论
3.1 物理化学性质表征
由XRD的表征结果可以看出(图1),用于制备GO的原料——石墨,在26.5°出现了石墨的(002)特征衍射峰(d(002)=0.34 nm),峰形尖锐,表明石墨原料晶体结构完整.相对石墨而言,GO的(002)衍射峰向低角度移至2θ=11.0°(d(002)=0.78 nm),石墨层间距的增大是由于氧化使石墨层间键合大量含氧官能团.RGO和N-RGO分别在24.5°(d(002)=0.381 nm)和26.2°(d(002)=0.342 nm)处左右出现了宽化的(002)衍射峰,显示出多层石墨烯结构的特点.而N-RGO相对RGO更小的(002)晶面间距,说明氮掺杂导致了更为密堆积的石墨烯基纳米层结构的形成,这与文献21中报道的氮掺杂对炭黑材料结构的影响结果类似.
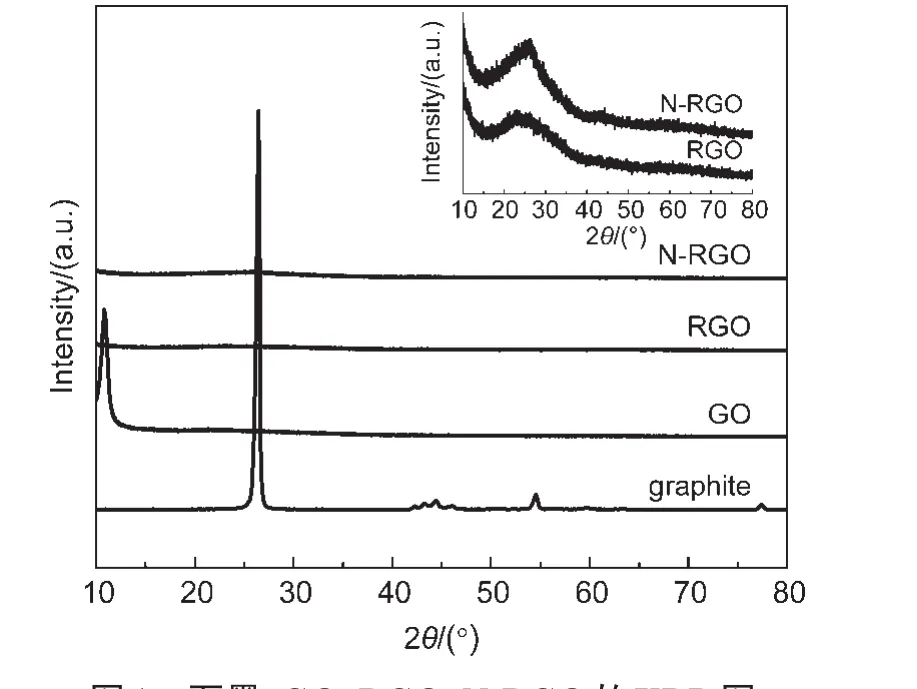
图1 石墨、GO、RGO、N-RGO的XRD图Fig.1 XRD patterns of graphite,GO,RGO,and N-RGO
拉曼技术是一种表征碳材料的有效手段.GO、RGO及N-RGO的拉曼表征结果如图2所示.从图中可知,GO、RGO、N-RGO的ID/IG值(D、G峰强度之比)依次增大(分别为0.83、0.89、0.99),这是因为在高温热解过程中,GO高温还原成RGO的过程造成的含氧官能团的分解,会在石墨烯片内产生大量的空穴缺陷,造成层内晶体颗粒的尺寸减小,使得ID/IG增大22(由0.83增加为0.89);而N-RGO的制备中GO的热解还原和氮的掺杂过程同时发生,氮掺杂会引起石墨烯表面缺陷程度的增加,导致了ID/IG由RGO的0.89增加至N-RGO的0.99.这种由氮掺杂引起的碳材料表面缺陷结构的增加有利于金属纳米粒子在碳材料表面的负载.23另一方面,RGO的拉曼图中在2700 cm-1附近出现了2D峰,说明热解还原生成的RGO是多层石墨烯结构.24同时我们可以看到,NRGO的拉曼图中几乎没有2D峰,这可能与氮掺杂引起石墨烯表面的缺陷增多,25以及石墨烯的层数进一步增加有关.
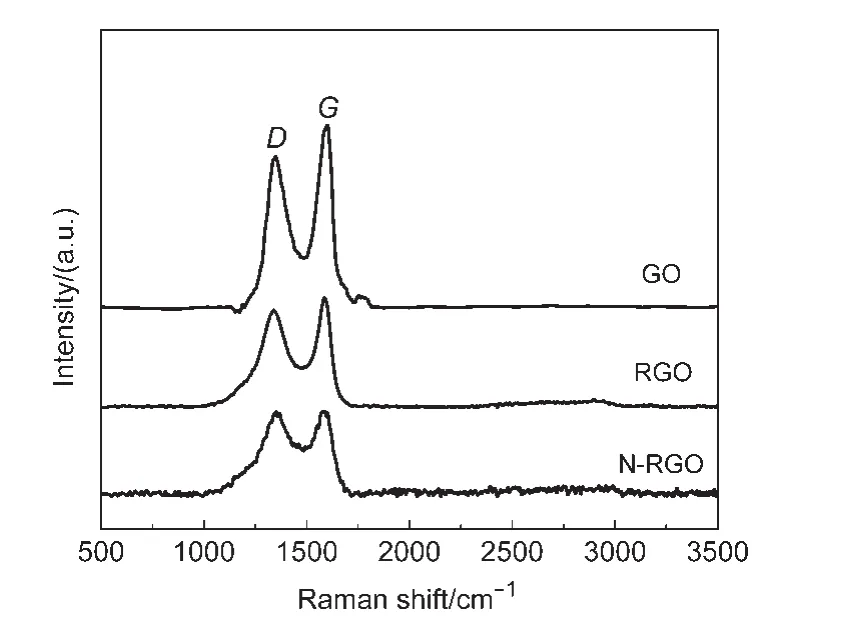
图2GO、RGO和N-RGO的拉曼光谱图Fig.2 Raman spectra of GO,RGO,and N-RGO
图3 为RGO、N-RGO与Pt/RGO、Pt/N-RGO的TEM表征.从图可以看出,热解还原得到的RGO呈现半透明的石墨烯片层结构,存在明显的褶皱.相对RGO来说,N-RGO样品的透明度及褶皱结构明显减少,显示出密堆积的层状结构,与XRD及Raman显示的结果一致.通过分析可知,在PANI-GO高温热解过程中,GO的热分解还原(形成RGO)、修饰在层状GO表面的聚苯胺的石墨化及对RGO的氮掺杂几乎同时发生,我们认为聚苯胺的石墨化过程很有可能是以GO和/或RGO为结构模板进行,聚苯胺形成的石墨化碳层插入到热解还原得到的RGO的层状结构中最终得到了密堆积层状结构的N-RGO.由Pt/RGO和Pt/N-RGO的TEM图(图3c和3d)可知,两个样品上Pt颗粒的尺寸大小接近,均为3-4 nm,但Pt/N-RGO上Pt颗粒的分布相对Pt/RGO更为均匀,这可能是因为N-RGO中碳晶格结构中掺杂的氮原子为Pt颗粒的形成提供了更多的成核中心.9
XPS是一种有效的表面化学组成分析手段,Pt/RGO及Pt/N-RGO的XPS全谱见图4.两个样品中都同时出现了C 1s,O 1s,Pt 4f及Pt 4d的信号.另外在Pt/N-RGO样品中还出现了N 1s的信号,表明氮成功掺杂进入了碳材料的结构(元素分析结果显示氮掺杂量为4.82%(原子分数)).Pt/N-RGO的N 1s信号显示为一对不对称的峰,表明N至少存在两种或两种以上的化学键构型.对N 1s进行分峰拟合结果显示,氮原子分别为吡啶型氮(398.1 eV)、吡咯型氮(399.8 eV)和石墨型氮(401.1 eV)的形式存在,含量分别为21.88%、6.97%、71.15%.可以看出Pt/N-RGO中的N大部分是以替代C原子的形式进入了石墨化碳层内形成石墨氮(71.15%),还有相当一部分N是以吡啶氮的形式存在于石墨化碳层的边缘结构(21.88%).这些类型的N物种作为富电子活性位有利于促进金属离子前驱体的还原和金属离子的良好分散,26并且对于加强活性组分Pt与载体之间的相互作用起到重要作用.27-29另外,我们还发现Pt/NRGO样品中O的含量为7.31%(原子分数),高于Pt/RGO样品中O的含量5.47%(原子分数),说明采用聚苯胺热解对石墨烯进行氮掺杂还会导致还原氧化石墨烯碳材料表面含氧官能团的增加.Pt/RGO及Pt/N-RGO的Pt 4f XPS谱分峰结果见图4(c,d).Pt 4f的XPS谱线通过分峰处理可以得到三对峰,分别对应于 Pt0、Pt2+和 Pt4+.Pt/RGO 样品中 Pt0(71.7、74.9 eV),Pt2+(72.8、76.2 eV),Pt4+(75.2、78.2 eV)的原子质量百分比含量分别为55.39%、26.19%、18.42%,而Pt/N-RGO样品中Pt0(71.6、74.8 eV),Pt2+(72.6、76.3 eV),Pt4+(75.2、78.2 eV)的原子质量百分比含量分别为:59.00%、26.43%、14.57%.Pt/N-RGO样品中Pt0含量明显高于Pt/RGO样品,表明N-RGO中N的供电子性质更有利于Pt前驱体的还原.31
3.2 甲醇电氧化性能测试
图5给出了Pt/N-RGO、Pt/RGO以及商业Pt/C(JM公司,Pt质量分数为20%)上CO溶出反应(图5(a))和甲醇电氧化反应(图5(b))的循环伏安曲线测试结果.可以看出Pt/N-RGO和Pt/C上CO溶出反应的起始电势(0.43 V)和峰电势(0.56 V)相当,低于Pt/RGO样品的相应值(分别为0.47和0.58 V),显示出比Pt/RGO更强的抗CO毒化能力.由Pt/N-RGO、Pt/RGO以及Pt/C的甲醇氧化循环伏安曲线可以看出,Pt/N-RGO和Pt/C电极催化剂上甲醇氧化的起始电势为0.20 V,明显低于Pt/RGO的相应值0.35 V;Pt/N-RGO在峰电势处Pt的质量比活性为165A·g-1,高于相应Pt/RGO样品的150 A·g-1,但低于商业Pt/C的180 A·g-1;另一方面,Pt/N-RGO在峰电势处Pt的面积比活性(本征活性)为3.67 A·m-2,明显高于相应的Pt/RGO样品及商业Pt/C(分别为2.13和2.28 A·m-2).
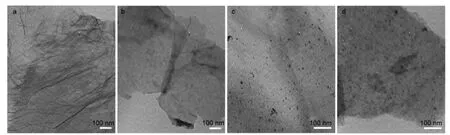
图3 RGO(a)、N-RGO(b)、Pt/RGO(c)和Pt/N-RGO(d)的TEM图Fig.3 TEM images of RGO(a),N-RGO(b),Pt/RGO(c),and Pt/N-RGO(d)

图4 Pt/RGO和Pt/N-RGO的XPS全谱图(a);Pt/RGO的Pt 4f谱图(b);Pt/N-RGO的N 1s XPS谱图(c)和Pt 4f XPS谱图(d)Fig.4 XPS survey spectra of Pt/RGO and Pt/N-RGO(a);Pt 4f XPS spectra of Pt/RGO(b);N 1s(c)and Pt 4f(d)XPS spectra of Pt/N-RGO
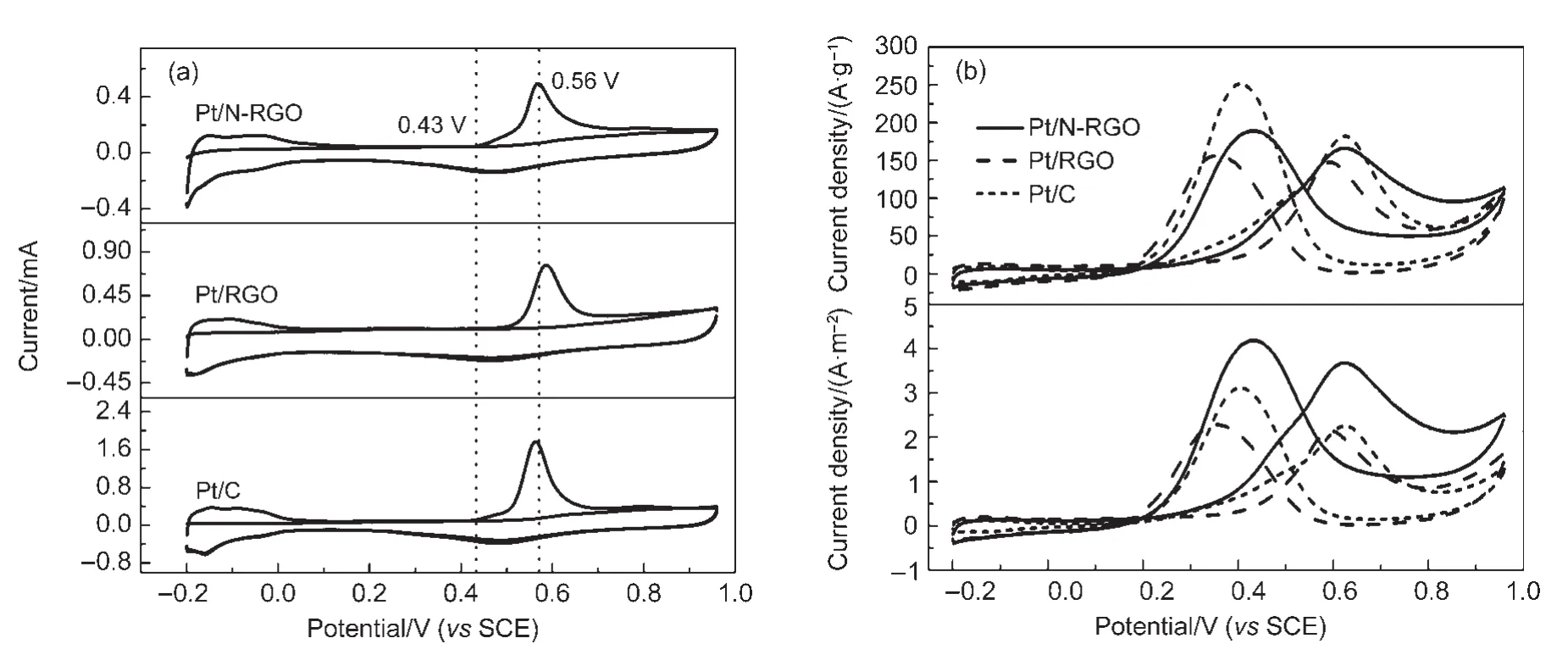
图5 Pt/N-RGO,Pt/RGO和Pt/C的CO溶出循环伏安图(a);Pt/N-RGO,Pt/RGO和Pt/C甲醇氧化的循环伏安图(b)Fig.5 CO stripping voltammograms(a)and cyclic voltammograms of the catalytic oxidation of methanol(b)for Pt/N-RGO,Pt/RGO and Pt/C
电极催化剂的稳定性是燃料电池得以应用的关键因素之一.Pt/N-RGO、Pt/RGO及Pt/C对甲醇电氧化反应催化稳定性测试结果如图6.其中,图6(a)为循环伏安曲线中正扫峰电流密度与扫描圈数的关系,图6(b)为0.40 V时的计时电流曲线.可以看到,在整个测试过程中Pt/N-RGO电极催化剂始终保持了比Pt/RGO和Pt/C更高的本征活性;经过300圈的循环伏安扫描及恒电势0.40 V下反应1 h后,Pt/N-RGO对甲醇电氧化反应的催化活性分别下降49%和40%,Pt/RGO的催化活性下降值则分别达到66%和83%,Pt/C的催化活性下降值分别为55%和47%,Pt/N-RGO的催化稳定性明显优于相应的Pt/RGO样品以及文献中报道的采用水热法等方法制备的N-RGO负载的Pt电催化剂.13以上研究结果表明,Pt/N-RGO电极催化剂样品对CO的抗毒化能力和甲醇氧化反应的催化活性及稳定性都明显优于相应的Pt/RGO样品.究其原因可能是Pt/N-RGO中含氮官能团的存在会使碳载体表面产生更多的缺陷(包括含氮及其含氧官能团造成的缺陷),有利于改善碳载体上负载的Pt颗粒的分散状态及提高Pt的抗CO毒化能力(如碳表面的含氧官能团有利于Pt上CO的氧化);31氮原子的供电子性质能够增加碳载体的导电性,并且进一步改变金属催化剂的电子结构(如Pt0的含量增加),32,33这些都有利于Pt/NRGO对甲醇电氧化反应催化活性和稳定性的提高.同时我们还可以看到,Pt/N-RGO催化甲醇电氧化反应的本征活性和稳定性都明显高于商业Pt/C催化剂,抗CO毒化能力与商业Pt/C催化剂相当.另外,我们将聚苯胺浓度增加一半,制备了N掺杂量更高的Pt/N-RGO样品,实验结果表明,该样品上Pt颗粒的分散程度降低(说明聚苯胺的浓度对N-RGO的特性及其负载的Pt颗粒的分散程度有明显影响),但对甲醇电氧化反应的本征活性仍明显优于相应的Pt/RGO样品.
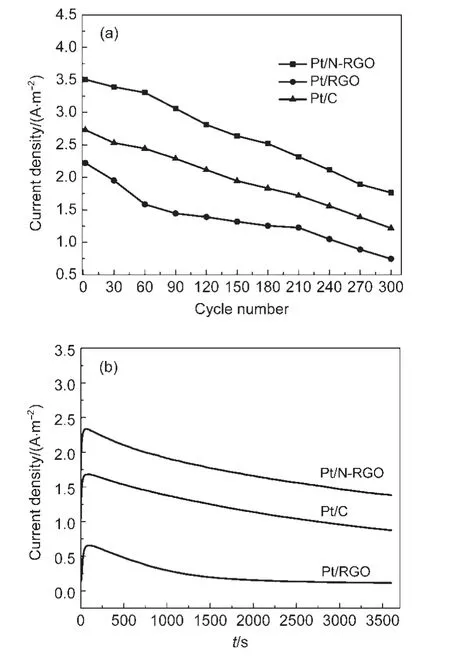
图6 Pt/N-RGO,Pt/RGO和Pt/C在甲醇电解液中正向扫描的氧化峰电流密度随扫描圈数变化的关系曲线(a);Pt/N-RGO,Pt/RGO和Pt/C计时电流曲线(b)Fig.6 Anodic peak current density for methanol oxidation plotting against cycle number on Pt/N-RGO,Pt/RGO and Pt/C electrodes catalysts(a);chronoamperometry of Pt/N-RGO,Pt/RGO,and Pt/C(b)
4 结论
采用高温热解聚苯胺修饰的氧化石墨烯(PANIGO)制备了氮含量为4.82%(原子分数)的氮掺杂还原氧化石墨烯碳材料N-RGO,并以其为载体负载Pt制备了Pt/N-RGO电极催化剂.与未掺杂氮的相应样品Pt/RGO相比,Pt/N-RGO表现出更强的抗CO毒化能力和更好的甲醇电氧化催化活性和稳定性.Pt/N-RGO更优的催化活性归因于氮掺杂改变了碳材料的表面缺陷程度及其负载的Pt电催化剂的电子结构,以及Pt-C之间的相互作用.以上结果表明,以聚苯胺为氮源制备的氮掺杂石墨烯基碳材料作为燃料电池电极催化剂的载体材料有一定的应用潜力.聚苯胺与氧化石墨烯的比例对N-RGO形貌及其负载Pt的分散程度和电催化性能的影响还有待进一步研究.
(1)Wei,D.;Liu,Y.;Wang,Y.;Zhang,H.;Huang,L.;Yu,G.Nano Lett.2009,9,1752.doi:10.1021/nl803279t
(2)Yang,Z.;Yao,Z.;Li,G.;Fang,G.;Nie,H.;Liu,Z.;Zhou,X.;Chen,X.;Huang,S.ACS Nano 2012,6,205.doi:10.1021/nn203393d
(3) Shao,Y.Y.;Sui,J.H.;Yin,G.P.;Gao,Y.Z.Appl.Catal.B 2008,79(1),89.doi:10.1016/j.apcatb.2007.09.047
(4) Xiong,B.;Zhou,Y.K.;O′Hayre,R.;Shao,Z.P.Appl.Surf.Sci.2013,266,433.doi:10.1016/j.apsusc.2012.12.053
(5)Wu,J.;Hu,F.;Hu,X.;Wei,Z.D.;Shen,P.K.Electrochimica Acta 2008,53(28),8341.doi:10.1016/j.electacta.2008.06.051
(6) Zhou,C.W.;Kong,J.;Yenilmez,E.;Dai,H.J.Science 2000,290,1552.doi:10.1126/science.290.5496.1552
(7) He,D.P.;Jiang,Y.L.;Lv,H.F.;Pan,M.;Mu,S.C.Applied Catalysis B:Environmental 2013,132-133,379.
(8) Xiao,X.;Zhou,Y.K.;Lu,J.M.;Tian,X.H.;Zhu,H.X.;Liu,J.G.Electrochimica Acta 2014,120,439.doi:10.1016/j.electacta.2013.12.062
(9)Zhang,L.S.;Liang,X.Q.;Song,W.G.;Wu,Z.Y.Phys.Chem.Chem.Phys.2010,12,12055.doi:10.1039/c0cp00789g
(10) Sun,L.;Wang,L.;Tian,G.G.;Tan,T.X.;Xie,Y.;Shi,K.Y.;Li,M.T.;Fu,H.G.RSC Adv.2012,2,4498.doi:10.1039/c2ra01367c
(11)Wang,Y.;Shao,Y.Y.;Matson,D.W.;Li,J.H.;Lin,Y.H.ACS Nano 2010,4,1790.
(12) Hassan,F.M.;Chabot,V.;Li,J.D.;Kim,B.K.;Ricardez-Sandoval,L.;Yu,A.P.J.Mater.Chem.A 2013,1,2904.
(13)Xu,X.;Zhou,Y.K.;Yuan,T.;Li,Y.W.Electrochimica Acta 2013,112,587.doi:10.1016/j.electacta.2013.09.038
(14)Lin,Z.Y.;Waller,G.;Liu,Y.;Liu,M.L.;Wong,C.P.Adv.Energy Mater.2012,2(7),884.
(15)Lin,Z.Y.;Song,M.K.;Ding,Y.;Liu,Y.;Liu,M.L.;Wong,C.P.Phys.Chem.Chem.Phys.2012,14,3381.doi:10.1039/c2cp00032f
(16) Sheng,Z.H.;Shao,L.;Chen,J.J.;Bao,W.J.;Wang,F.B.;Xia,X.H.ACS Nano 2011,5,4350.doi:10.1021/nn103584t
(17)Lin,Z.Y.;Waller,G.;Liu,Y.;Liu,M.L.;Wong,C.P.Nano Energy 2013,2,241.doi:10.1016/j.nanoen.2012.09.002
(18) Lai,L.F.;Potts,J.R.;Zhan,D.;Wang,L.;Poh,C.K.;Tang,C.H.;Gong,H.;Shen,Z.X.;Lin,J.Y.;Rodney,S.R.Energy Environ.Sci.2012,5,7936.doi:10.1039/c2ee21802j
(19)Wu,G.;Mack,N.H.;Gao,W.;Ma,S.G.;Zhong,R.Q.;Han,J.T.;Baldwin,J.K.;Zelenay,P.ACS Nano 2012,6(11),9764.doi:10.1021/nn303275d
(20)Hummers,W.S.;Offeman,R.E.J.Am.Chem.Soc.1958,80,1339.doi:10.1021/ja01539a017
(21) Wu,G.;Swaidan,R.J.;Li,D.Y.;Li,N.Electrochimica Acta 2008,53,7622.doi:10.1016/j.electacta.2008.03.082
(22) Stankovicha,S.;Dikina,D.A.;Piner,R.D.;Kohlhaas,K.A.;Kleinhammes,A.;Jia,Y.Y.;Wu,Y.;Nguyen,S.B.T.;Ruoff,R.S.Carbon 2007,45(7),1558.doi:10.1016/j.carbon.2007.02.034
(23) Liu,S.;Wang,J.;Zeng,J.;Ou,J.;Li,Z.;Liu,X.;Yang,S.G.J.Power Sources 2010,195(15),4628.doi:10.1016/j.jpowsour.2010.02.024
(24) Ferrari,A.C.;Meyer,J.C.;Scardaci,V.;Casiraghi,C.;Lazzeri,M.;Mauri,F.;Piscanec,S.;Jiang,D.;Novoselov,K.S.;Roth,S.;Geim,A.K.Phys.Rev.Lett.2006,97,187401.doi:10.1103/PhysRevLett.97.187401
(25)Kudin,K.N.;Ozbas,B.;Schniepp,H.C.;Prud′homme,R.K.;Aksay,I.A.;Car,R.Nano Lett.2008,8,36.doi:10.1021/nl071822y
(26) Xin,Y.C.;Liu,J.G.;Zhou,Y.;Liu,W.M.;Gao,J.;Xie,Y.;Yin,Y.;Zou,Z.G.Electrochimica Acta 2012,60,354.doi:10.1016/j.electacta.2011.11.062
(27) Kuo,P.L.;Chen,W.F.;Huang,H.Y.;Chang,I.C.;Dai,S.A.J.Phys.Chem.B 2006,110,3071.
(28)Wu,G.;Li,D.;Dai,C.;Wang,D.;Li,N.Langmuir 2008,24,3566.doi:10.1021/la7029278
(29) Groves,M.N.;Chan,A.S.W.;Malardier,J.C.;Jugroot,M.Chem.Phys.Lett.2009,481,214.doi:10.1016/j.cplett.2009.09.074
(30) Zhou,Y.;Neyerlin,K.;Olson,T.S.;Pylypenko,S.;Bult,J.;Dinh,H.N.;Gennett,T.;Shao,Z.P.;O'Hayre,R.Energy Environ.Sci.2010,3(10),1437.doi:10.1039/c003710a
(31)Wang,S.Y.;Jiang,S.P.;Wang,X.;Guo,J.Electrochimica Acta 2011,56,1563.doi:10.1016/j.electacta.2010.10.055
(32) Zheng,S.F.;Hu,J.S.;Zhong,L.S.;Wan,L.J.;Song,W.G.J.Phys.Chem.C 2007,111,11174.doi:10.1021/jp0727042
(33)Zheng,B.;Zheng,W.T.;Zhang,K.;Wen,Q.B.;Zhu,J.Q.;Meng,S.H.;He,X.D.;Han,J.C.Carbon 2006,44,962.doi:10.1016/j.carbon.2005.10.009
猜你喜欢
化工管理(2022年14期)2022-12-02
中国化肥信息(2022年3期)2022-05-05
昆钢科技(2022年1期)2022-04-19
纺织科学研究(2021年7期)2021-08-14
理化检验-化学分册(2020年5期)2020-06-15
石油石化绿色低碳(2019年6期)2019-02-13
中国化肥信息(2016年27期)2016-05-17
通信电源技术(2016年6期)2016-04-20
中国塑料(2015年7期)2015-10-14
中国塑料(2014年11期)2014-10-17
