
FenO(n=3~13)团簇稳定结构和电磁特性的DFT计算
2014-09-13 07:25:44韩辉云王荣娥贾惠琳
石家庄职业技术学院学报 2014年2期
韩辉云, 王荣娥, 贾惠琳
(1.河北师范大学 汇华学院,河北 石家庄 050091;2.衡水市第十三中学 高二物理组,河北 衡水 053000;3.河北师范大学 物理科学与信息工程学院,河北 石家庄 050024)
铁,是柔韧且有延展性的银白色金属,其含量在地壳中排名第四,在宇宙中排名第九.铁元素在元素周期表中是第26号元素,其原子量为56,位于第四周期第VIII族,外围的电子轨道排布是3d64s2,是过渡金属.在材料科学的研究领域中,纳米团簇,特别是过渡金属团簇,是科学家们热心研究的课题之一,他们与块体材料不同,都呈现出反常的磁学性质.铁作为3d过渡金属的典型,其团簇有着极其重要的应用.如,在磁学方面,量子计算机硬件上使用的最佳磁性存储材料就是Fe8团簇[1].铁被广泛应用于许多工业的催化过程中,如作为合成单壁碳纳米管的催化剂与合成氨的催化剂[2-3].由于以上原因,铁团簇一直是理论与试验研究的热点.
20世纪90年代中期以来,人们对于铁氧团簇的试验研究逐渐增加.这是因为铁和氧的混合团簇在表面催化和高密度磁记忆存储器等方面具有广泛的应用.文献[4]通过光电子能谱对电子结构进行了系统的研究,发现对于铁氧小团簇而言,氧原子的多少影响团簇电子的亲和力.文献[5]使用离子束质谱仪研究了铁氧正离子团簇,发现阳离子在整个团簇中占主导地位.文献[6]利用密度泛函理论(density functiond theory,缩写为DFT)研究了环状、塔状与鼓状的铁氧中性团簇的性质.文献[7]通过基于DFT的第一性原理的分子动力学方法研究了FenOm(n=1~5)团簇的结构和磁性,发现氧原子数目与磁性有关系.文献[8]研究了(Fe2O3)n的笼状和非笼状团簇,发现(Fe2O3)n的笼状结构具有高稳定性.文献[9]在广义局域自旋密度泛函理论基础上用关于非线性磁性结构的平面波赝势法研究了FeOn和FeOn负离子团簇的电子结构和特性.文献[10]采用二项式方案构建了FenOm(n+m=4)团簇的大量可能初始结构,运用广义梯度近似(generalized gradient approximation,缩写为 GGA)及PW91交换关联泛函对这些初始结构进行了优化和频率分析,得到12个稳定的异构体.在此基础上计算和分析了它们的结合能、对称性、键长、磁矩、最高占据分子轨道(the highest occupied molecular obital,缩写为 HOMO)和最低未占据分子轨道(the lowest unoccupied molecular orbital,缩写为 LUMO)的能隙,发现Fe-O键在FenOm(n+m=4)阳离子团簇的稳定中具有重要作用,团簇的总磁矩主要取决于铁原子的磁矩和各个原子磁矩排布的情况.文献[11]对Fe2O3阳离子团簇结构进行了具体的计算、分析,通过对其最初的60个结构的优化得到28个不同的异构体,并对这些稳定的异构体进行结合能、HOMO-LOMO和磁矩的分析,同分异构体的总磁矩主要取决于有磁性的铁原子在这些团簇上的磁性排列,电荷的转移对这些异构体也有重要的影响.同铝一样,铁表面也会受到空间环境中大量存在的原子氧的氧化腐蚀,因此,铁团簇表面原子氧的吸附研究对于冶金工业有一定的参考价值.为此,本文系统研究了氧原子掺杂后的铁团簇FenO(n=3~13)的结构演化、电子特性和磁矩.
1 计算方法
本课题组基于先前对过渡金属铁团簇Fen(n=2~13)研究的结果并参考有关铁氧掺杂团簇的研究文献[4-12],构造尽可能多的单个氧原子掺杂纯铁团簇的初始结构,以便于寻找到近基态结构.使用基于DFT的科学计算程序Dmol3对建成的初始构型进行结构优化,在优化过程中,采用GGA的交换关联密度泛函PW91和双数值极化函数基组(doublenumberical basis plus polarized functions,缩写为DNP),实空间的球形轨道截断半径取5.0Å.在自洽计算的过程中,总的能量收敛标准为10-6Hatree,最大的作用力收敛标准为10-3Hartree/Å,原子最大的位移收敛标准为10-3Å.团簇中的电荷与自旋计算采用 Mülliken布居分析.采用DIIS(direct inversion in iterative subspace)方法,可加快自洽场的收敛速度.
为了证明参数的可靠性,我们计算了Fe2O2的电离势,并与文献[13]的试验结果进行了对比:本文计算框架下得到的Fe-O键长为1.808Å,电离势为7.852eV;文献[13]得到的电离势为7.020eV.可见结果与试验值符合较好,在允许的范围内,说明该参数适合用于研究铁氧团簇.
2 计算结果与讨论
2.1 FenO(n=3~13)团簇的低能态结构
利用上面介绍的计算方案,本文对所有的FenO(n=3~13)团簇的可能构型进行了优化,获得了一系列的最稳态结构以及亚稳态结构.
对于Fe3O团簇,优化得到了两种低能态结构:三角锥型是最低的能量结构,团簇中的氧原子加在Fe3团簇三角形的上端;具有C2v对称性的四边形平面结构,比基态能量高0.320eV.
对于Fe4O团簇,基态结构是氧原子位于类蝴蝶形Fe4团簇上的C2v结构;其余分别是在类三角锥型的Fe4团簇的顶端加入一个氧原子得到的对称性为C3v的双金字塔结构,比基态结构高0.475eV;在平面Fe4团簇的不同位置添加一个氧原子,得到一个具有Cs对称性结构和一个C1对称性的平面结构,与它们的基态结构相比,其能量分别高出0.734eV和0.508eV.
在Fe5O团簇中,于双金字塔型的Fe4团簇的不等价位置加入一个氧原子得到两个锥形结构.其基态结构具有C4v对称结构;具有Cs对称结构的另一种结构不稳定,结合能比最稳态高0.210eV.其余低能态结构分别是:在Fe5团簇的四角锥型结构上戴帽一个氧原子得到的C2v结构,其基态能量比此时结构的能量低0.391eV;在平面五边形Fe5团簇上戴帽一个氧原子得到C5v结构,其能量比基态能量高2.374eV.
对于Fe6O团簇,经过一系列的几何优化后,得到七个同分异构体,都接近于基态.其中,基态结构具有C3v对称结构且结合能为-26.036eV,这个结构由一个Fe6三棱锥上戴帽一个氧原子得到,其余的六个低能态结构对称性分别为Cs,C5v,D4h和D6h;在类三棱锥上戴帽一个氧原子得到的Cs对称性结构的能量比基态能量高0.578eV;同样一种具有Cs对称性结构的Fe6O,由Fe5O一种结构的侧边添加一个铁原子而得到;氧原子位置在四棱锥中心的Fe6O的一种D4b对称结构的能量高出基态能量4.144eV.D6h,C5v团簇是在碗状Fe6上戴帽一个氧原子得到的,其能量分别比基态能量高5.488eV和2.696eV.
对于Fe7O,得到八个同分异构体.其中,具有C3v对称性的双锥结构是基态结构,此结构是在Fe6O基态结构的底端戴帽一个铁原子得到的.其余结构为:在类三角锥的两条不等价边分别添加一个氧原子得到两种结构;在Fe6碗状结构上戴帽一个氧原子得到;在Fe6O最后一种同分异构体结构底端加上一个铁原子而得到;在Fe6双三角锥结构的不等价位置添加氧原子可得到对称性分别为Cs和C2v的两种结构.还有一种结构由飞碟状的Fe6上戴帽一个氧原子而得到,其能量高出基态能量0.641eV.
Fe8O所得到的同分异构体有六个,具有四种对称结构,它们分别为C2v,C4v,D4d和D6h.具有C2v对称结构的分别为:具有类笼状结构的基态结构,它是在Fe7O一种结构底端添加一个铁原子而形成的;另一种则是在塔状铁团簇内嵌一个氧原子得到的,其能量比基态能量高2.426eV.其余结构分别是:在十面体上戴帽氧原子得到;在正六面体上戴帽氧原子得到;在十面体中心添加一个氧原子得到,其能量比基态能量高3.817eV;具有D6h对称结构的最后一种结构则是由飞碟状Fe9用氧原子取代中心位置的一个铁原子得到的,其能量高于基态能量2.319eV.
对于Fe9O团簇,优化得到了五个低能态结构.基态结构是中空笼状结构,对称结构为C4v;剩下的五个同分异构体的对称性分别是C2,D3h,C2v,C4v和Cs,他们都是中空笼状结构.
经过优化后的Fe10O团簇,其最稳定结构中氧原子位于笼状团簇的中心位置,对称结构为D4d;对称结构分别为C2v,C3v和C4v的四种同分异构体,氧原子都位于笼状结构的顶端;具有D2d和D4d对称结构的两种结构中,氧原子位于中心位置;还有一种结构可以描述为在Fe11笼状团簇中心替代一个氧原子而得到;最后一种双锥形团簇结构由氧原子替代而形成.
Fe11O团簇的几何优化结果显示,其最稳态结构是具有C2v对称性的笼状结构,其余的异构体中氧原子都位于笼状的表面,对称性分别为C5v和C3v.
相比于Fe11O团簇,Fe12O中的基态结构具有C2v对称性;对于对称性为C5v的两种结构分别为在正圆锥体和斜笼状团簇上戴帽氧原子得到,它们的能量比基态能量高1.696eV和2.398eV.
在Fe13O团簇中经过结构优化,得到七个同分异构体,所有的低能态结构的氧原子都戴帽于笼状结构:对于同为C4v对称性的三种结构中,在变形的十四面体上戴帽一个氧原子的结构是最稳态结构,而另两个则是在正十四面体和十七面体上戴帽氧原子形成,其能量分别比基态能量高0.442eV和0.618eV;对称性为C6v的一种结构为在正五边柱形结构上戴帽氧原子得到,其与基态能量相比高出5.084eV;相对于笼状结构,另外两种是类盘状结构,其中一个是在盘状顶端戴帽一个氧原子得到的,而另一个则可以描述为氧原子代替碗状团簇中的中心铁原子而得到,其能量比基态能量高3.091eV.
表1为FenO(n=3~13)团簇中计算基态结构的结果,它显示了团簇的对称性、团簇结合能Eb、HOMO-LUMO能隙 H-L gap(eV)、氧原子在混合团簇中所带的电荷和团簇的总磁距.
总之,在n≤13的铁氧混合团簇中,除了Fe10O团簇以外,其他掺杂团簇中的氧原子多位于团簇的对称轴上,且多数倾向于团簇的表面.相比单一的团簇,混合团簇的性质发生了改变.

表1 FenO(n=3~13)团簇中计算基态结构的结果
2.2 结果与分析
2.2.1 相对稳定性
团簇的平均每原子结合能定义为相互作用原子间的总能与相应自由原子体系的总能之差除以团簇包含的原子总数.
图1给出了FenO团簇的平均结合能随尺寸的变化规律,为了比较,同时给出了Fen的结果.纯Fe团簇基态结构的平均结合能大体上随着团簇原子数的增长保持单调递增趋势,说明在生长过程中,纯Fe团簇能够继续获得能量,只是在原子数为11时出现了明显的谷值,说明Fe11具有比相邻团簇较低的稳定性.而FenO(n=3~13)团簇局域谷值出现在n等于10和12时.在n等于6,9和11时,都分别具有比相邻团簇更高的稳定性.FenO(n=3~13)团簇每一原子的平均结合能比相同原子数下的纯Fen团簇的每一原子平均结合能高,掺杂O原子能够加强纯Fe团簇的稳定性.

图1 FenO团簇和Fen+1团簇平均每原子结合能随尺寸的变化情况
图2显示的是,在FenO团簇基态结构中Fe-Fe平均键长和Fe-O平均键长,以及纯Fen+1团簇基态结构中的Fe-Fe平均键长随着团簇尺寸的变化规律.纯铁团簇的Fe-Fe平均键长基本呈现弱增长趋势.FenO团簇中的Fe-Fe平均键长随着团簇尺寸的增加,呈现出大幅度的震荡.
团簇的幻数和其相对应的电子壳层结构是研究团簇的一个重要方面.团簇结合能的二阶差分可用△2E(n)=Eb(n+1)+Eb(n-1)-2Eb(n)来计算.
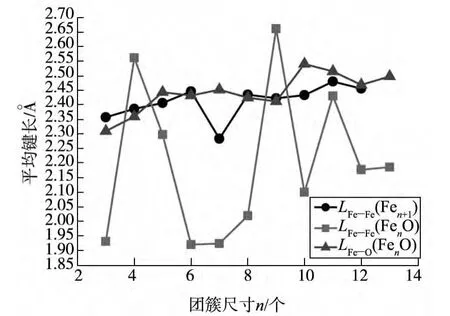
图2 Fen+1和FenO团簇中的Fe-Fe,Fe-O平均键长随团簇尺寸的变化情况
图3显示了FenO(n=3~13)团簇的总结合能的二阶差分能.与纯Fe团簇相比,对于FenO团簇而言,局域峰值出现在n=6,9,11时,这就暗示这些团簇比相邻团簇的稳定性高.因此,铁氧团簇中的幻数团簇是Fe6O,Fe9O和Fe11O.值得注意的是,FenO团簇中的Fe10O团簇的二阶差分能出现了较大的谷值,此团簇不稳定性.在纯Fe团簇中,二阶差分能的峰值出现在n=6,8,10时.
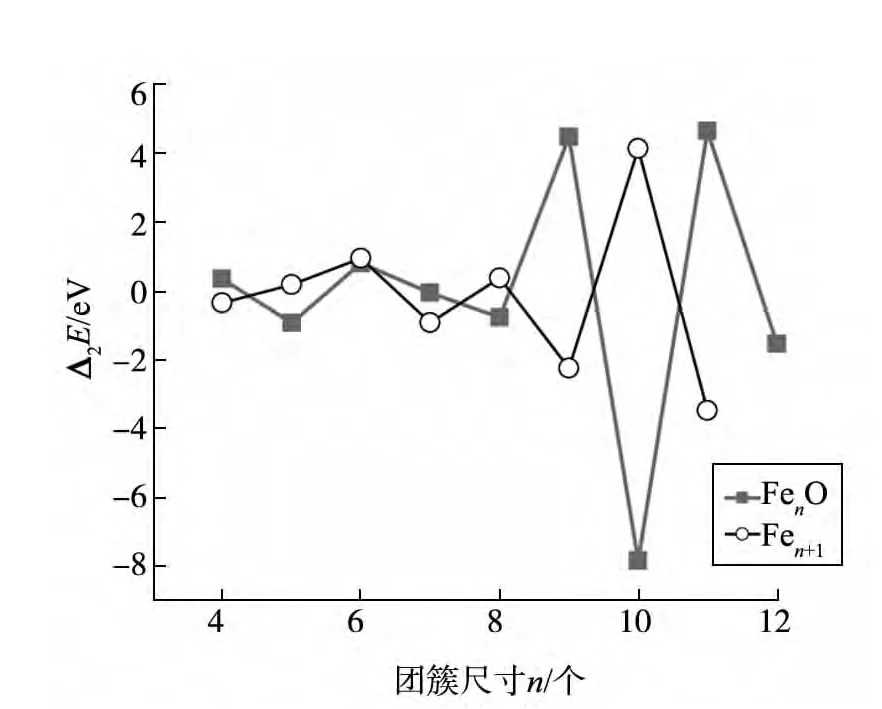
图3 Fen+1和FenO团簇结合能的二阶差分能随团簇尺寸n的变化情况
2.2.2 电子结构和磁特性
基于分子轨道理论,原子轨道所构成的线性组合就是该体系的轨道.团簇或者分子体系的电子填充也是由低能到高能,这和原子外层电子的分布类似.在化学反应中,能量最高的占据分子轨道HOMO,最低的占据分子轨道LUMO,这与化学键的形成或断裂有密切关系.HOMO与LUMO之间的能隙决定于化学反应能力的强弱.图4描绘的是团簇FenO和Fen+1(n=3~13)团簇基态结构中团簇尺寸变化引起的能隙变化情况.可以看出,与纯铁团簇的能隙震荡幅度相比,氧原子的加入加剧了团簇能隙大小的震荡;铁氧团簇的峰值出现在n=6,9,13时,表明Fe6O,Fe9O和Fe13O这三个团簇有较高的化学稳定性;在n=5,8,12时,HOMOLUMO能隙出现谷值,表明Fe5O,Fe8O和Fe12O团簇具有较强的化学活性,更容易发生化学反应.

图4 FenO和Fen+1(n=3~13)团簇的基态结构中H-L能隙随团簇尺寸n的变化情况
图5显示的是FenO(n=3~13)团簇最低能态结构中的Mülliken电荷分布情况.可以看出,在n=3~13的范围内,O原子的电荷转移随着团簇的原子数呈现震荡行为,震荡幅度大约在-0.680eV到-0.743eV之间.在n=6,9和12的团簇中,O原子具有相对较高的电荷转移量,证实了团簇有相对高的稳定性,例如,Fe6O团簇中O原子的电荷转移大于Fe5O和Fe7O中O原子的电荷转移,说明Fe6O团簇中Fe和O的静电作用比Fe5O和Fe7O中的Fe-O间的静电作用强,所以Fe6O团簇为幻数团簇.在所有的FenO团簇中,O原子都带有负电荷,说明电子都从Fe原子转移到O原子,Fe原子为施主,而O原子为电子的受主.值得关注的是,从Fe9O到Fe10O团簇的电荷转移量出现了很大的缩减.在FenO(n=3~13)团簇中,除Fe10O外,基态结构中氧原子均为表面原子,电荷转移量一般大于0.680eV,而氧原子在团簇中心的Fe10O的电荷转移量是0.617eV,这表明电荷转移数目可能与团簇的基态几何结构有一定的关系.
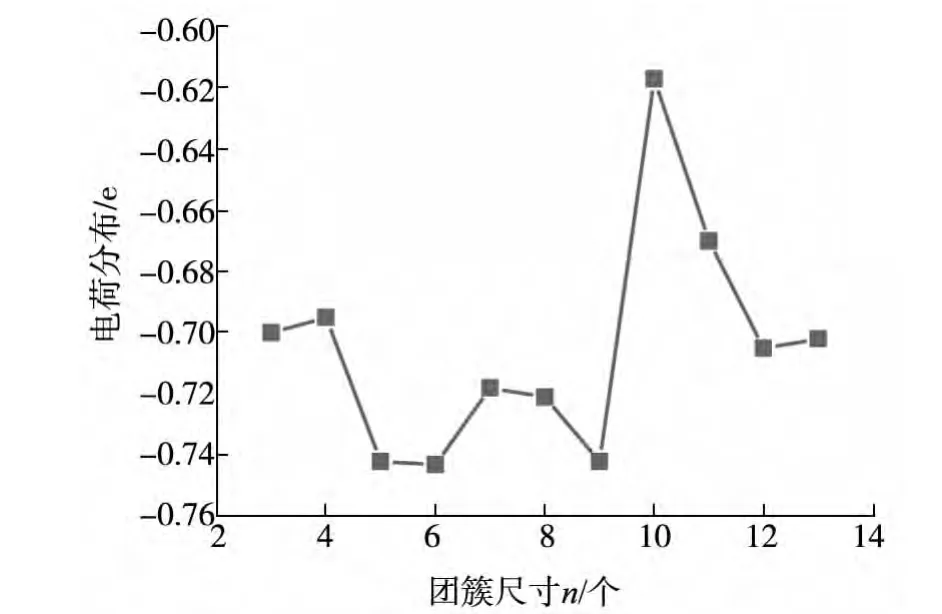
图5 FenO(n=3~13)团簇基态中氧原子的Mülliken电荷随团簇尺寸n的变化情况
在团簇物理中,磁特性是一个有趣的现象之一,它有不同于分子或者块体材料的特征.本文根据已经优化好的最稳态结构,计算了FenO(n=3~13)团簇的总磁矩,在图6中画出了团簇的总磁矩随团簇尺寸n的变化情况.随着团簇尺寸n的增加,团簇磁距总体上呈上升趋势,但n=5,10时除外.将图5和图6进行对比可以发现,在n=10处,O原子的Mülliken电荷转移出现了绝对的减少,Fe10O的磁矩也出现了很大的谷值.Fe10O团簇是唯一一个氧原子位于团簇中心位置的团簇.FenO(n=3~13)团簇的磁距可能与氧原子的掺杂位置和铁原子到氧原子的电荷转移有一定的关联.

图6 FenO(n=3~13)团簇的总自旋磁矩随着团簇尺寸n的变化情况
3 结论
基于DFT的电子结构,采用GGA下的PW91交换关联函数,系统地分析了FenO团簇基态结构的几何结构、结合能、能隙和磁距.研究发现,除Fe10O外,FenO(n=3~13)团簇中氧原子的位置总是处于团簇的表面;相比于纯铁团簇,铁氧团簇的结构稳定性和化学惰性增强;随着氧原子的加入,团簇Fe6O,Fe9O,Fe11O为幻数团簇;在FenO(n=3~13)团簇中,总磁距与氧原子的掺杂位置和电荷转移有一定的关系.
[1]MICHSEL N L,LOSS D.Quantum Computing in Molecular Magnets[J].Nature,2001,410:789-793.
[2]IIJIMA S,ICHIHASHI T.Single-shell Carbon Nanotubes of 1nm Diameter[J].Nature,1993,363:603-605.
[3]BETHUNE D S,KLANG C H,DE VRIES M,et al.Cobalt-catalysed Growth of Carbon Nanotubes with Single-atomic-layer Walls[J].Nature,1993,363:605-607.
[4]WANG Lan-sheng,WU Hong-bin,DESAI S.Sequential Oxygen Atom Chemisorption on Surfaces of Small Iron Clusters[J].Phys Rev Lett,1996,76:4853-4956.
[5]GRIFFIN J B,ARMENTROUT P B.Guided Ion Beam Studies of the Reactions of Fen+(n=2~18)with O2:Iron Cluster Oxide and Dioxide Bond Energies [J].J Chem Phys,1997,106:4448-4462.
[6]JONES N O,REDDY B V,KHANNA S N,et al.Structural Growth in Iron Oxide Clusters:Rings,Towers,and Hollow Drums[J].Phys Rev B,2005,72:165411(1-4).
[7]SHIROISHI H,ODA T,HAMADA I,et al.Structure and Magnetism on Iron Oxide Clusters FenOm(n=1~5):Calculation from First Principles[J].Eur Phys J D,2003,24:85-88.
[8]DING Xue-lei,XUE Wei,MA Yan-ping,et al.Density Functional Study on Cage and Moncage(Fe2O3)nClusters[J].J Chem Phys,2009,130:014303(1-8).
[9]GUTSEV G L,KHANNA S N,RAO B K,et al.Electronic Structure and Properties of FeOnand FeOn-Clusters [J].J Phys Chem A,1999,103:5812-5822.
[10]徐本富,杨佳路,童小菲,等.FenOm+(n+m=4)团簇的构型、电子结构特征和磁性 [J].物理学报,2010,59:7845-7849.
[11]XU Ben-fu,YANG Chuan-lu,WANG Mei-shan,et al.The Geometric Structure and Electronic Properties of Fe3O3+Clusters[J].Phys.B,2011,406:200-204.
[12]MA Qing-min,XIE Zun,WANG Jing,et al.Structures,Binding Energies and Magnetic Moments of Small Iron Clusters:A Study Based on All-electron DFT [J].Solid State Comm,2007,142:114-119.
[13]LI Ming,LIU Shu-rong,ARMENTROUT P B.Collision-induced Dissociation Studies of FemOn+:Bond Energies in Small Iron Oxide Cluster Cations,FemOn+(m=1~3,n=1~6)[J].J Chem Phys,2009,131:144310(1-16).
猜你喜欢
物理化学学报(2024年4期)2024-07-18 00:00:00
无机化学学报(2023年2期)2023-02-27 03:29:26
发明与创新·中学生(2023年2期)2023-01-09 03:50:05
椰城(2021年12期)2021-12-10 06:08:52
创新作文(小学版)(2019年27期)2019-11-30 05:48:14
物理学报(2017年21期)2017-11-10 08:25:38
Zoological Research(2017年4期)2017-08-24 07:50:09
小学生必读(低年级版)(2016年3期)2016-11-11 06:12:15
腐蚀与防护(2016年7期)2016-09-14 09:30:56
数学大王·趣味逻辑(2016年1期)2016-01-29 09:57:29
