
机械力对血管内皮细胞一氧化氮合成的调控机制及致动脉粥样硬化作用
2014-09-13 01:36:20李庆杨漪
中国老年学杂志 2014年23期
李 庆 杨 漪
(石家庄市中医院功能科,河北 石家庄 050051)
血流机械力变化导致的动脉粥样硬化主要在于它破坏了血管内皮细胞和平滑肌细胞的一氧化氮(NO)生成的系统平衡〔1〕。本文重点讨论血流机械力〔剪切力和环壁张力对内皮细胞NO的生成和NO合酶(NOS)〕表达的调控机制,并强调其介导血管内皮细胞的感染、氧自由基的产生及最终引发血管壁重建(主要是平滑肌细胞参与)的机制。
1 剪切力对血管内皮细胞NO生成及NOS表达的影响
1.1剪切力影响血管内皮细胞生成NO 单向的剪切力通过两个机制影响血管内皮细胞生成NO。在剪切力开始作用后几秒,NOS受刺激开始产生NO,与此同时,剪切力产生了一个短暂而强烈的细胞内钙离子浓度的提高,随之,高浓度的钙调蛋白捆绑到NOS上并增强NOS的活性。当胞内钙离子浓度提高到最大值后返回到剪切力刺激前的正常水平,NO的生成却不断提高,也就是说剪切力诱导的NO生成并不依赖于细胞内钙离子浓度的改变。这个独立的作用机制来源于NOS的特别位点的磷酸化作用,即磷酸化导致了电子从NOS的还原区域向酶的加氧区域移动〔2〕。磷酸化特别位点在NOS的第635和1 177个氨基酸(均为丝氨酸,由蛋白激酶A介导)〔3〕。几小时以后,剪切力引发了NOS mRNA转录和相应的蛋白合成增加,结果是内皮细胞产生大量的NO〔4〕。这个NOS的上调表达包括两个过程,一个是短暂的NOS mRNA转录的提高,一个是稳定的持续的NOS mRNA高表达〔5〕,前一个转录提高的信号转导通路主要是先刺激络氨酸激酶cSrc,而后再刺激Ras,Raf,MEK 1/2和Erk 1/2四个通路;后一个稳定过程主要是依赖cSrc通路,没有丝裂原活化蛋白激酶(MAP)激酶通路参与。
2 剪切力对NOS mRNA转录的影响
剪切力刺激NOS mRNA 转录仅仅持续1 h左右。这个现象将会有巨大的生理学意义。如,简单的身体锻炼会提高人体内的NOS产量,进一步说,NOS mRNA短暂的高爆发表达类似一个药物负荷剂量的作用,即通过提高 mRNA 稳定性,一个稳定状态的NOS mRNA高表达将会持续出现。剪切力对NOS mRNA转录的调节主要是超激活NOS 刺激物(顺势作用元件),这个刺激物在转录起始位点上游-984到-990之间〔6〕〔此区域包含剪切力反应序列5-¢CCAGAG-3¢〔7〕,超激活通过核因子(NF)κB结合到此位点来实现〕。此剪切力特别反应序列的突变将导致NOS 刺激物对剪切力刺激失去反应。NF-κB存在于细胞质,为一不活跃的三聚体(P50、P65、IκB三个独立功能的活动区域)。当抑制性子亚单位IκB被其上游的激酶所磷酸化时〔8〕,它便与P65、P50分离,而后P65、P50依次转位到细胞核,在这与相关基因的顺势作用元件同源物结合。有实验表明,在NOS mRNA激活过程中,剪切力引发了P65、P50转位并提高了二者的迁移速度,在NOS基因的顺势作用元件上存在剪切力反应序列,P65、P50将结合此序列上来完成上述剪切力刺激反应。值得强调的是,在NF-κB对NOS表达的影响过程中,NF-κB的激活将会提高与感染相关的基因的表达,这些基因包括血管内皮细胞黏附分子(VCAM)1、细胞内黏附分子(ICAM)1、组织因子、单核细胞趋化因子(MCP)1。上述感染相关基因参与了动脉粥样硬化的形成似乎与NF-κB刺激NO生成而有抗动脉粥样硬化形成的作用矛盾。最近研究显示存在一个负反馈机制调节了剪切力依赖的NF-κB激活NO的瞬时生成过程〔9〕。相关实验显示、向内皮细胞体外补充NO将减弱剪切力对eNOS基因表达的促进作用及降低NOS表达刺激物的活性;相反,向内皮细胞体外补充NOS抑制剂将起到促进的作用。除了激活NF-κB,剪切力同时也提高介导NO生成的相关酶的活性。如前所述,NF-κB的激活导致P50/P65二聚体转位到细胞核,在这里二聚物结合到NOS基因的顺式作用元件而提高NOS mRNA的转录和NOS蛋白合成。P50的亚硝化作用将抑制其转位〔10〕,以此限制NF-κB在细胞核内的活性,这个机制说明了是P50的亚硝化作用导致剪切力刺激下的NO大量生成返回到刺激前的水平。这个负反馈机制能用如下的实验说明,用L-NG-硝基精氨酸甲酯抑制NO的生成将导致瞬间的P50向核内转位,而转位不受剪切力刺激的影响,相反,用突变的P50基因(相关蛋白缺乏亚硝化作用位点)转染内皮细胞,内皮细胞会产生大量的NOS表达。无论内皮细胞是否会受到剪切力刺激,这个高表达不会减弱。以上结果说明在剪切力激活NF-κB、提高NOS表达、促进NO生成之间存在一个负反馈通路。在基本的非病理生理状态下,正常水平的NOS蛋白决定了剪切力依赖的NOS基因顺式作用元件的超激活的时间。如果NOS蛋白处在一个高水平状态,在剪切力刺激后不久它就能够催化生成足够数量的NO来终止NF-κB的激活作用。如果NOS蛋白处在一个较低水平,NF-κB的活性将持续保持在一定水平,NOS mRNA的转录活性也会不断提高,这足以使NOS恢复并保持在正常水平。在不正常的病理生理状态下,这个负反馈的抑制作用方面将会改变,此时剪切力刺激会变成一个病理性的炎性刺激〔11〕。例如,NOS基因促进物的多态性能够抑制剪切力依赖的NOS的转录,同理,在剪切力刺激下,NOS辅助因子四氢生物蝶呤(BH)4的缺乏能够抑制NO生成和导致NF-κB活性持续保持。某些内生的NOS抑制物,例如非对称的二甲基精氨酸,能够抑制NO生成和不断促进NF-κB的核转位(在剪切力刺激下)。这些作用能够导致一个剪切力刺激下持续内皮细胞感染的病理现象(而不是剪切力刺激下的抑制内皮细胞感染)。这些机制充分说明了剪切力刺激的NO生成是抑制内皮细胞感染的关键因素,而在缺乏NO状态下,剪切力刺激却是一个致感染因素。
2 环壁张力对血管内皮细胞NO的生成及NOS表达的影响
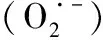
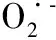
2.4内皮细胞内ROS含量的影响因素 NOS缺乏的高血压小鼠体内ROS含量降低,暗示NADPH氧化酶和脱耦联的NOS是血管内ROS不断提高的重要源泉。虽然NOS缺乏的高血压小鼠体内ROS含量明显降低,但仍然保持一些,这个事实暗示在鼠内皮细胞和平滑肌细胞内,还有一些其他的生成ROS的资源,它包括黄嘌呤氧化酶、线粒体、细胞色素p450等,一个实验已证实〔21〕,在NADPH氧化酶缺乏的小鼠内皮细胞内,ROS的生成没有降低,而其对肿瘤坏死因子(TNF)-α和佛波醇酯的刺激反应降低。
2.5环壁张力与内皮细胞依赖的血管舒张机制的关系 研究提示〔22〕,在血管高张刺激下,NOS的脱耦联也是血管内过氧化氢(H2O2)产物不断生成的重要源泉,同理,抑制BH4的合成将导致内皮细胞依赖的血管舒张。有实验〔22〕证实,在高血压小鼠体内,eNOS酶的辅因子BH4氧化将导致BH4含量降低。
两个机制导致了H2O2刺激血管舒张,一个机制是H2O2刺激过氧化氢酶并使之与鸟苷酸环化酶结合形成复合物〔23〕,随之复合物刺激环磷酸腺苷大量生成。另一个机制是H2O2是一个内皮细胞依赖的超极化因子〔24〕。相比其他氧化物,H2O2是相对稳定的,能从一个细胞扩散到另一个细胞,在细胞内H2O2也有靶目标,因此,H2O2对血管平滑肌细胞的增生和肥大起着极其重要的作用。
3 参考文献
1Pan S.Molecular mechanisms responsible for the atheroprotective effects of laminar shear stress〔J〕.Antioxid Redox Signal,2009;11:1669-82 .
2Rafikov R,Fonseca FV,Kumar S,etal.eNOS activation and NO function:structural motifs responsible for the post translational control of endothelial nitric oxide synthase activity〔J〕.J Endocrinol,2011;210:271-84.
3Boo YC,Sorescu G,Boyd N,etal.Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms:role of protein kinase A〔J〕.J Biol Chem,2002;277:3388-96.
4Kawai Y,Yokoyama Y,Kaidoh M,etal.Shear stress-induced ATP-mediated endothelial constitutive nitric oxide synthase expression in human lymphatic endothelial cells〔J〕.Am J Physiol Cell Physiol,2010;298:C647-55.
5Moore JP,Weber M,Searles CD.Laminar shear stress modulates phosphorylation and localization of RNA polymerase Ⅱ on the endothelial nitric oxide synthase gene〔J〕.Arterioscler Thromb Vasc Biol,2010;30:561-7.
6Davis ME,Grumbach IM,Fukai T,etal.Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding〔J〕.J Biol Chem,2004;279:163-8.
7Esnick N,Collins T,Atkinson W,etal.Platelet-derived growth factor B chain promoter contains a cis-acting fluid shear-stress-responsive element〔J〕.Proc Natl Acad Sci USA,1993;90:7908.
8Davis ME,Cai H,Drummond GR,etal.Shear stress regulates endothelial nitric oxide synthase expression throughc-Src by divergent signaling pathways〔J〕.Circ Res,2001;89:1073-80.
9Grumbach IM,Chen W,Mertens SA,etal.A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription〔J〕.J Mol Cell Cardiol,2005;39:595-603.
10Matthews JR,Botting CH,Panico M,etal.Inhibition of NF-kappaB DNA binding by nitric oxide〔J〕.Nucleic Acids Res,1996;24:2236-42.
11Cattaruzza M,Guzik TJ,Slodowski W,etal.Shear stress insensitivity of endothelial nitric oxide synthase expression as a genetic risk factor for coronary heart disease〔J〕.Circ Res,2004;95:841-7.
12Landmesser U,Dikalov S,Price SR,etal.Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension〔J〕.J Clin Invest,2003;111:1201-9.
13Arora S,Vaishya R,Dabla PK,etal.NAD(P)H oxidases in coronary artery disease〔J〕.Adv Clin Chem,2010;50:65-86.
14Tanner FC,Meier P,Greutert H,etal.Nitric oxide modulates expression of cell cycle regulatory proteins:a cytostatic strategy for inhibition of human vascular smooth muscle cell proliferation〔J〕.Circulation,2000;101:1982-9.
15Weiss EP,Fontana L.Caloric restriction:powerful protection for the aging heart and vasculature〔J〕.Am J Physiol Heart Circ Physiol,2011;301:H1205-19.
16Cabral PD,Garvin JL.Luminal flow regulates NO and O2(-) along the nephron〔J〕.Am J Physiol Renal Physiol,2011;300:F1047-53.
17Lo J,Patel VB,Wang Z,etal.Angiotensin-converting enzyme 2 antagonizes angiotensin Ⅱ-induced pressor response and NADPH oxidase activation in Wistar-Kyoto rats and spontaneously hypertensive rats〔J〕.Exp Physiol,2013;98:109-22.
18Koba S,Watanabe R,Kano NO,etal.Oxidative stress exaggerates skeletal muscle contraction-evoked reflex sympathoexcitation in rats with hypertension induced by angiotensin Ⅱ〔J〕.Am J Physiol Heart Circ Physiol,2013;304:H142-53.
19Laursen JB,Somers M,Kurz S,etal.Endothelial regulation of vasomotion in apoE-deficient mice:implications for interactions between peroxynitrite and tetrahydrobiopterin〔J〕.Circulation,2001;103:1282-8.
20Patel KB,Stratford MR,Wardman P,etal.Oxidation of tetrahydrobiopterin by biological radicals and scavenging of the trihydrobiopterin radical by ascorbate〔J〕.Free Radic Biol Med,2002;32:203-11.
21Li JM,Mullen AM,Yun S,etal.Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α〔J〕.Circ Res,2002;90:143-50.
22Cosentino F,Barker JE,Brand MP,etal.Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice〔J〕.Arterioscler.Thromb Vasc Biol,2001;21:496-502.
23Norton CE,Jernigan NL,Kanagy NL,etal.Intermittent hypoxia augments pulmonary vascular smooth muscle reactivity to NO:regulation by reactive oxygen species〔J〕.J Appl Physiol,2011; 111:980-8.
24Matoba T,Shimokawa H,Nakashima M,etal.Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice〔J〕.J Clin Invest,2000;106:1521-30.
猜你喜欢
橡塑技术与装备(2022年10期)2022-10-03 07:39:14
水利科技与经济(2021年11期)2021-12-04 09:11:32
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
中国眼镜科技杂志(2019年9期)2019-11-11 12:15:32
生物学通报(2019年3期)2019-02-17 18:03:58
安徽医科大学学报(2016年12期)2017-01-15 14:21:48
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
中国病理生理杂志(2015年8期)2015-12-21 12:38:16
云南中医学院学报(2014年5期)2014-07-31 18:00:10
