
应用Gibson Assembly方法构建植物表达载体
2014-08-31 07:17:04戢志呈江雁翔刘耀光张群宇
华南农业大学学报 2014年5期
戢志呈,江雁翔,刘耀光,张群宇
(亚热带农业生物资源保护与利用国家重点实验室/华南农业大学 生命科学学院,广东 广州 510642)
应用Gibson Assembly方法构建植物表达载体
戢志呈,江雁翔,刘耀光,张群宇
(亚热带农业生物资源保护与利用国家重点实验室/华南农业大学 生命科学学院,广东 广州 510642)
【目的】构建方便快捷的植物表达载体.【方法】应用Isothermalinvitrorecombination system or “Gibson Assembly”方法设计包含片段间20~25 bp互补重叠序列的引物,通过PCR扩增出带有首尾重叠的目的DNA片段,可将1个或多个片段和线性化的载体一步组装成表达载体.【结果和结论】 利用该方法快速构建了多个水稻基因的全长以及部分缺失编码区表达载体.此外,还通过对Gibson Assembly连接产物进行PCR扩增后再连接到载体,提高多DNA片段组装的效率.本方法不受目的片段内部限制性酶切位点的限制,可广泛应用于各种载体的构建.
Gibson Assembly; 体外重组; 植物表达载体构建
表达载体是研究目的基因功能和遗传工程的重要工具,在亚细胞定位、目的基因表达、基因敲除、酵母双杂交和单杂交、T-DNA插入突变等技术方法中广泛应用.传统载体的构建基于限制性内切酶和T4DNA连接酶的方法,它要求先从细胞或组织中分离目的基因的DNA片段,然后利用限制性内切酶切断双链DNA分子从而产生黏性末端或平末端.如果线性化载体也产生相同的末端,二者可以在T4DNA连接酶作用下重新形成磷酸二酯键,构成一个完整的DNA分子[1].此方法操作容易、但针对多个片段的连接,则需要进行多次的酶切连接,而且还受限于可利用的酶切位点.
Gibson Assembly方法是Gibson等[2]发明的将多个DNA片段在1次反应中实现分子间连接.此方法关键要求是连接片段与线性化载体首尾有一小段的互补重叠序列.利用T5核酸外切酶(T5 exonuclease)的5′→3′外切酶活性产生黏性末端,带有互补的重叠序列DNA配对,再利用Phusion DNA polymerase 补齐片段上的缺口部分,最后通过TaqDNA Ligase将多个DNA片段连接起来.2009年Gibson等[3]应用Gibson Assembly方法设计40 bp的互补重叠序列合成了3个5 kb的片段,并且成功地插入到1个8 kb的细菌人工染色体上.2010年Gibson等[4]以600个含有60 bp互补重叠序列的片段利用Gibson Assembly方法采用三轮组装的方法成功合成了16.3 kb的小鼠线粒体基因.Jiang 等[5]利用Gibson Assembly方法开发了一步法构建植物干涉载体的技术:从植物基因组中扩增出目标片段,一步法形成反向互补的发夹结构,并同时连接入干涉载体中.
本研究应用Gibson Assembly方法构建了1个水稻转录因子基因Os05g0144300的全长及其4个缺失突变植物转化载体.另外,将从不同来源得到的启动子PNos、目的基因ATP5-N和orfH79及终止子TNos这4个表达元件,一步法构建了水稻红莲型细胞质雄性不育基因orfH79表达载体.本研究可为Gibson Assembly方法应用于植物表达载体的构建提供借鉴.
1 材料与方法
1.1 试验材料
植物材料:水稻‘中花11’和红莲型不育系‘粤泰A’ ,哥伦比亚型拟南芥.载体和菌株:pER8载体,pCAMBIA1300载体,大肠埃希菌DH10B均为华南农业大学遗传工程实验室保存.
1.2 Gibson Assembly方法反应体系
反应体系根据Gibson Assembly方法[6]和Jiang等[5]改良方法配制.2× IR Buffer(Enzyme-reagent mix): 320 μL 5× IR Buffer, 0.3 μL T5 exonuclease (10 U/μL) (Epicentre), 20 μL Phusion polymerase (2 U/μL) (NEB), 160 μLTaqligase (40 U/μL) (NEB), 加ddH2O至0.8 mL,分装为5 μL的等份,-20 ℃条件下储存.
1.3 Os05g0144300基因全长及缺失突变体载体的构建
Os05g0144300基因全长及缺失突变载体构建引物见表1.分别以水稻‘中花11’叶片抽提的基因组DNA和cDNA为模板PCR扩增Os05g0144300基因的启动子和目的基因cDNA片段.PCR反应体系为:50 μL反应体系中加入2×PCR Buffer for KOD FX 25 μL,2 mmol/L dNTPs 10 μL,100 μmol/L Primer F 0.15 μL,100 μmol/L Primer R 0.15 μL,KOD FX 1 μL,模板1 μL,灭菌去离子水12.7 μL.PCR反应条件为:94 ℃预变性2 min; 98 ℃变性10 s,56 ℃退火30 s,68 ℃延伸3 min,30个循环;68 ℃延伸10 min.PCR产物电泳检测,采用回收试剂盒(TaKaRa公司)进行纯化回收后,加入10% 体积的3 mol/L NaAc和2.5倍体积的无水乙醇,-80 ℃放置3 h,10 000 r/min 4 ℃离心沉淀,0.3 mol/L的TE溶解PCR产物,并置于-20 ℃条件下保存.
启动子扩增的上下游引物为P-EcoRⅠF和P-EcoRⅠR.目的基因1~1 128氨基酸(aa)片段上下游引物为CDS-F、1 128 aaR,1~730 aa片段的上下游引物为CDS-F、730 aaR,1~869aa片段的上下游引物为CDS-F、869 aaR,1~443 aa片段的上下游引物为CDS-F、440 aaR,731~1 128 aa片段的上下游引物为731~1 128 aaF 、1 128 aaR,1~741 aa片段的上下游引物为CDS-F、741 aaR,870~1 128 aa片段的上下游引物为870~1 128 aaF、1 128 aaR.
用EcoRⅠ酶切质粒pCAMBIA1300,酶切50 μL反应体系:5 μL Buffer、12.5 μL质粒、0.5 μL内切酶、32 μL ddH2O,37 ℃酶切1.5 h后终止反应.酶切产物凝胶回收,纯化,再乙醇沉淀纯化.Gibson Assembly连接体系为10 μL:载体3 μL,克隆片段1 μL,混合酶6 μL,50 ℃反应20 min.连接产物电击转化DH10B,用质粒抽提试剂盒(TaKaRa公司)抽取阳性克隆质粒DNA进行EcoRⅠ酶切鉴定,并送上海美吉公司进行测序,测序正确后命名为PCP.
用SalⅠ对PCP质粒进行酶切,线性化后的载体与1~1 128 aa片段、1~730 aa片段 、1~869 aa片段用Gibson Assembly方法分别连接(方法同上).成功连接后质粒分别命名为PCP-1、PCP- 2、 PCP-3.线性化后的载体与1~443 aa片段、731~1 128 aa片段混合连接,成功连接后质粒命名为PCP- 4.线性化后的载体与1~741 aa、870~1 128 aa混合连接,成功连接后的质粒命名为PCP-5.

表1 Os05g0144300基因全长及缺失突变体载体引物Tab.1 Primers for a vector construction of the full-length and truncated Os05g0144300 gene
1) 引物序列中小写字母碱基为引物的互补重叠序列.
1.4 orfH79表达载体的构建
orfH79表达元件组装相关引物见表2.以pER8载体质粒为模板,PNos-F、PNos-R为引物扩增PNos,TNos-F、TNos-R为引物扩增TNos[7].以水稻红莲型‘粤泰A’叶片的 cDNA为模板,orfH79-F、orfH79-R为引物扩增orfH79 (GeneBank accession NO. AB254027.1)[8].以拟南芥叶片的cDNA为模板,ATP5N-F、ATP5N-R为引物扩增ATP5的线粒体定位信号肽序列ATP5-N(At5g13450)[9].
PNos 、ATP5-N、orfH79和TNos这4个片段用Gibson Assembly方法进行一次性连接,连接体系和连接条件同上.连接产物连接到T载体pMD®18-T(TaKaRa公司)转化大肠埃希菌,以阳性克隆质粒为模板,扩增orfH79各表达元件进行PCR验证并测序.

表2 orfH79表达元件扩增引物
1) 小写字母为引物的互补重叠序列.
2 结果与分析
2.1 Os05g0144300基因的全长和缺失突变载体的构建
为了研究水稻转录因子Os05g0144300基因的功能,利用PCR从水稻‘中花11’的基因组DNA扩增出该基因编码区上游的启动子约3 000 bp,同时用EcoRⅠ酶切线性化pCAMBIA1300载体,采用Gibson Assembly方法将二者连接.成功连接的载体命名为PCP,EcoRⅠ酶切验证结果显示插入片段与目标分子大小一致 (图1).
根据Os05g0144300蛋白质功能域预测结果,设计Os05g0144300基因全长和缺失突变表达载体(图2).
从水稻‘中花11’ cDNA扩增出Os05g0144300基因全长1~1 128 aa片段3 384 bp 、1~730 aa片段2 100 bp、1~869 aa片段2 607 bp、1~443 aa片段1 329 bp、731~1 128 aa片段1 194 bp、1~741 aa片段2 223 bp、870~1 128 aa片段777 bp.用SalⅠ酶切线性化PCP质粒,将线性化的载体分别与1~1 128 aa片段、1~730 aa片段、1~869 aa片段连接,线性化的载体与1~443 aa片段、731~1 128 aa片段混合连接,线性化的载体与1~741 aa片段、870~1 128 aa片段混合连接,连接均采用Gibson Assembly方法进行.获得的5个重组质粒PCP-1、PCP- 2、PCP-3、PCP- 4、PCP-5经限制性内切酶SalⅠ酶切鉴定,结果显示其大小一致(图3).
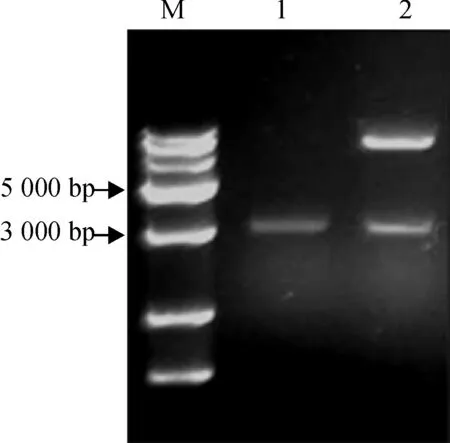
M: DNA marker DL15000;1:Os05g0144300基因启动子片段的扩增;2:PCP质粒酶切.
图1 PCP载体限制性内切酶EcoRⅠ酶切检测结果
Fig.1 Electrophoresis results of PCP plasmid digested byEcoRⅠrestriction enzyme
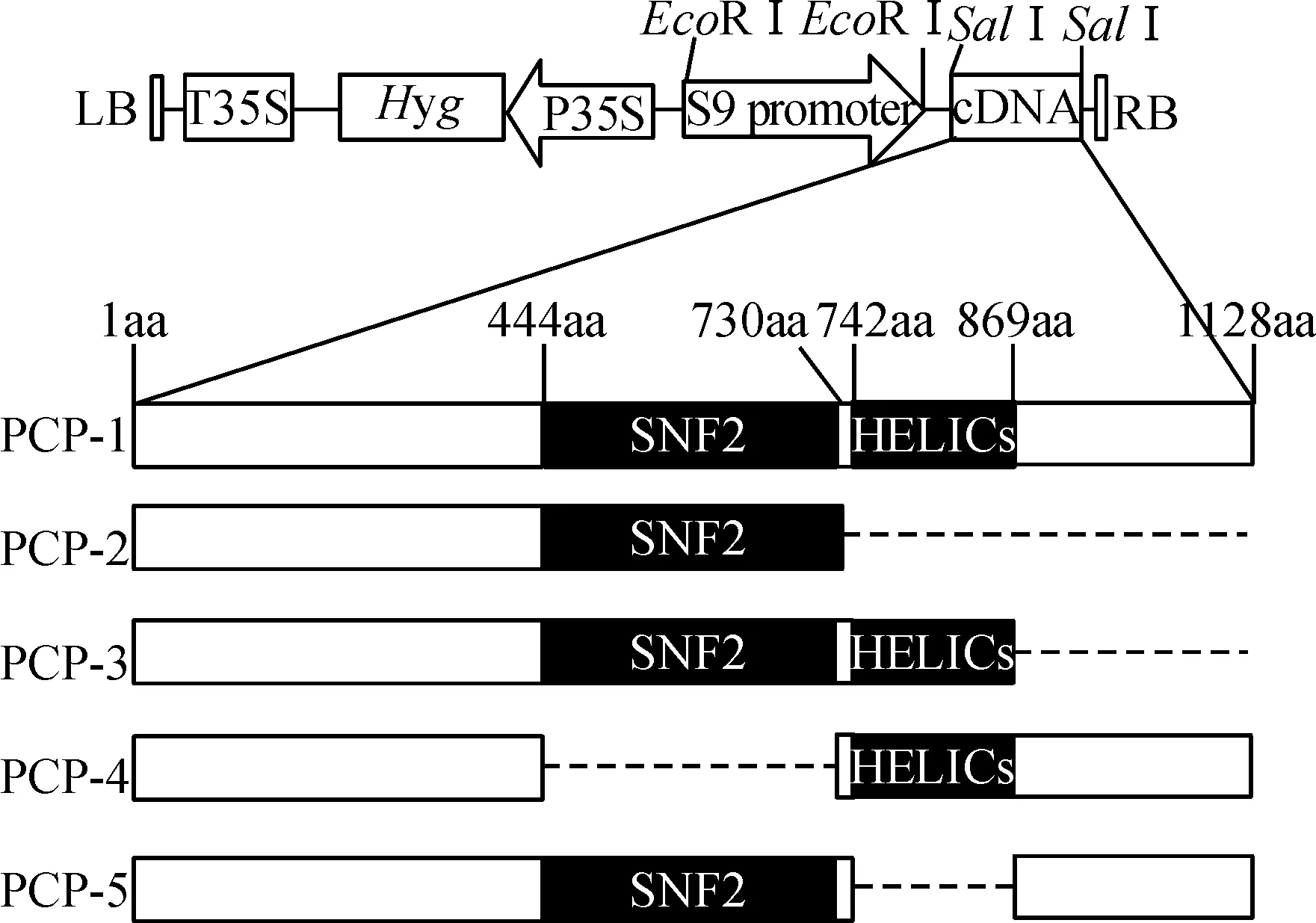

图2Os05g0144300基因全长结构和缺失突变载体示意图
Fig.2 Schematic diagram of the construction of full-length and truncated rice geneOs05g0144300 expression vectors
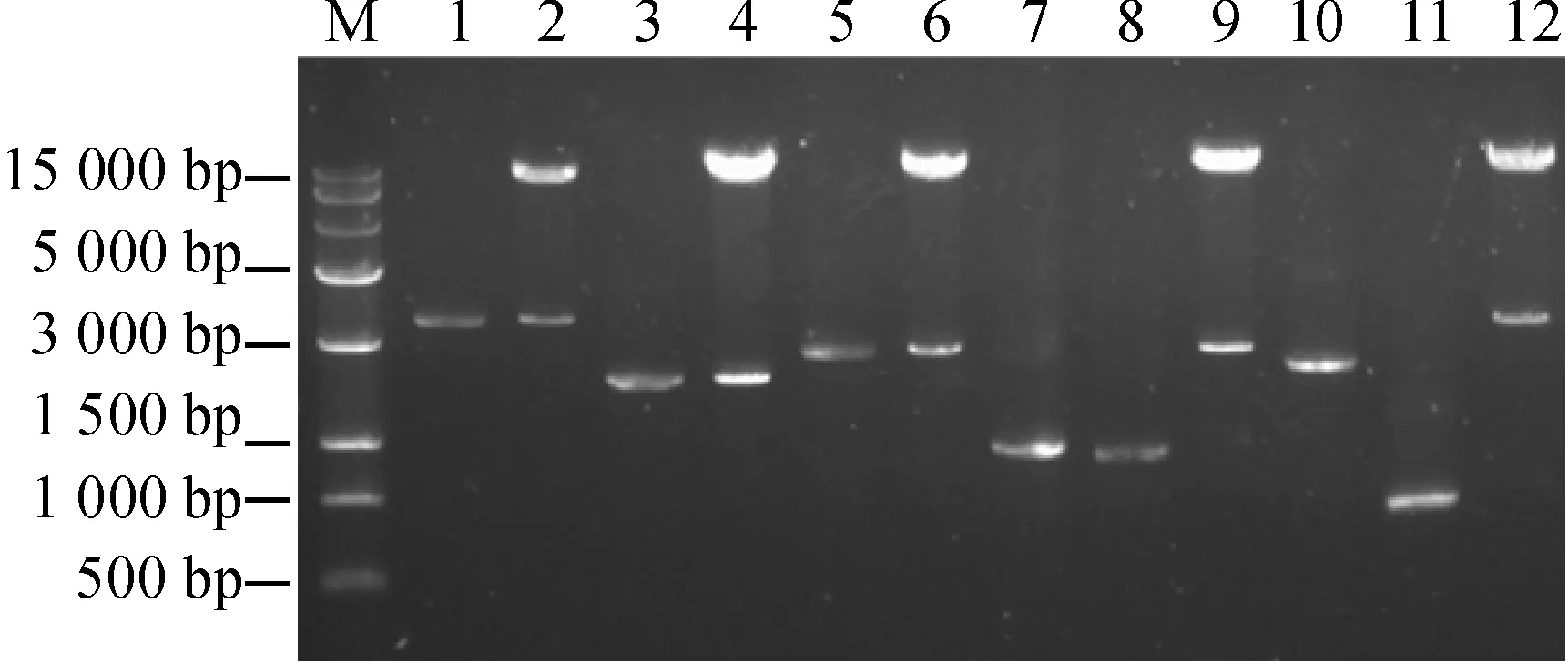
M: DNA marker DL15000;1:1~1 128 aa;2:PCP-1酶切;3:1~730 aa;4:PCP- 2酶切;5:1~869 aa;6:PCP-3酶切;7:1~443 aa;8:731~1 128 aa;9:PCP- 4酶切;10:1~741 aa;11:870~1 128 aa;12:PCP-5酶切.
图3Os05g0144300基因全长和缺失突变载体限制性内切酶SalⅠ酶切检测结果
Fig.3 Electrophoresis results of plasmids of the full-length and truncated rice geneOs05g0144300 expression vectors digested bySalⅠ restriction enzyme
2.2 orfH79表达元件载体的构建
利用PCR从pER8载体扩增出PNos片段333 bp、TNos片段275 bp,从拟南芥的cDNA扩增出ATP5基因N端序列354 bp,从水稻红莲型不育系‘粤泰A’ 的cDNA扩增出orfH79基因598 bp.PNos、ATP5-N、orfH79、TNos这4个片段,用Gibson Assembly方法进行连接.鉴于多个DNA片段一次性连接到载体的方法虽然可以得到阳性克隆,但效率较低.我们对以上4个DNA片段利用Gibson Assembly方法进行连接后,取部分连接产物先进行一次PCR扩增,扩增产物命名为PAHT(约1 600 bp),再将PAHT连接到TA载体或植物表达载体上转化大肠埃希菌(图4).这种改良可以大大提高阳性克隆比率.挑取12个单菌落进行菌落PCR鉴定,结果显示全部为阳性克隆.以阳性克隆质粒为模板,从中扩出orfH79各个表达元件以及PAHT,片段大小与预期相符(图5).
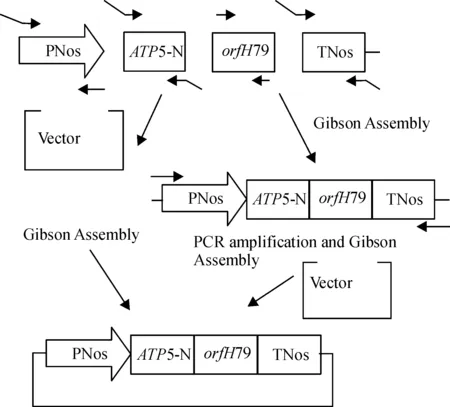
图4 orfH79表达元件载体构建示意图
Fig.4 Schematic diagram of the construction oforfH79 expression vector

M:DNA marker DL2000;1:PNos;2:ATP5-N;3:orfH79;4:TNos;5:PAHT.
图5orfH79表达元件载体各片段和组装的完整表达框的PCR检测
Fig.5 Detections of assembling fragments of theorfH79 expression vector by PCR
3 讨论
传统的载体构建基于酶切连接重组的经典克隆方法,但是该种方法在针对一些较长片段或多个DNA序列时,载体可供选择的限制性酶切位点往往受到限制[10].采用Gibson Assembly方法仅需要对载体进行单酶切线性化,利用互补DNA序列可实现多段DNA序列的无缝连接,不需要依赖特定的酶切位点.本方法与传统酶切连接方法相比有以下优点:①传统酶切连接方法需要较长的连接反应时间,而本方法只需温浴20~30 min后即可转化;②传统酶切连接方法每次只能连接1个插入片段,多个片段的组装需要逐次进行限制性内切酶酶切连接反应,随着连接片段数目的增多,载体的多克隆位点将无法满足插入片段的要求.而本方法只需对载体进行一种限制性内切酶酶切,获得线性化载体,而插入片段不需要进行酶切产生相同的黏性末端,即对多克隆位点的要求很低;③减少了试验操作步骤,由于插入片段的连接依赖于互补片段,而不是限制性内切酶酶切位点的黏性末端,减少了插入片段的酶切纯化等步骤;④该方法的高效性还体现在对多个不同目的片段的克隆上:当需要用同一个载体对多个基因进行克隆时,仅需要在合成引物时在特异引物的末端加上载体线性化位点两侧的序列,而不需要分析目的片段的限制性内切酶酶切位点等信息,即可以工厂化进行多个载体的构建;另外,在实际的Gibson Assembly方法应用中,一次反应连接多个DNA片段时可能会出现效率下降,阳性克隆少的问题,我们对其进行改良,对多片段的连接产物进行1次PCR扩增后再连接到载体,大大提高了阳性克隆率.
应用Gibson Assembly方法,克隆片段的长度可以是几百碱基,也可以达到900 kb[3],甚至可以用于人工合成细菌的全基因组[11-12].Gibson Assembly方法已经应用于动物[4]、酵母基因组的克隆[2, 13-14].本研究应用该方法成功构建了Os05g0144300基因全长及缺失突变植物转化载体和水稻红莲型不育系‘粤泰A’细胞质雄性不育基因orfH79表达元件载体,为同类植物表达载体的快速构建提供了方法借鉴.
[1] SMITH H O, WILCOX K W. A restriction enzyme fromHemophilusinfluenzae: I: Purification and general properties [J]. J Mol Biol, 1970, 51(2): 379-391.
[2] GIBSON D G. Synthesis of DNA fragments in yeast by one-step assembly of overlapping oligonucleotides [J]. Nucleic Acids Res, 2009, 37(20): 6984- 6990.
[3] GIBSON D G, YOUNG L, CHUANG R Y, et al. Enzymatic assembly of DNA molecules up to several hundred kilobases [J]. Nat Methods, 2009, 6(5): 343-345.
[4] GIBSON D G, SMITH H O, HUTCHISON C R, et al. Chemical synthesis of the mouse mitochondrial genome [J]. Nat Methods, 2010, 7(11): 901-903.
[5] JIANG Yanxiang, XIE Min, ZHU Qinlong, et al. One-step cloning of intron-containing hairpin RNA constructs for RNA interference via isothermalinvitrorecombination system [J]. Planta, 2013,238(2): 325-330.
[6] GIBSON D G. Enzymatic assembly of overlapping DNA fragments [J]. Methods Enzymol, 2011(498): 349-361.
[7] SOLAIMAN D K, SOMKUTI G A. Shuttle vectors developed fromStreptococcusthermophilusnative plasmid [J]. Plasmid, 1993, 30(1): 67-78.
[8] ZHANG Hong, LI Shaoping, YI Ping, et al. A Honglian CMS line of rice displays aberrant F0 of F0F1-ATPase [J]. Plant Cell Rep, 2007, 26(7): 1065-1071.
[9] ROBISON M M, LING X, SMID M P, et al. Antisense expression of mitochondrial ATP synthase subunits OSCP (ATP5) and gamma (ATP3) alters leaf morphology, metabolism and gene expression inArabidopsis[J]. Plant Cell Physiol, 2009, 50(10): 1840-1850.
[10]张亚旭. DNA重组技术的研究综述 [J]. 生物技术进展, 2012,2(1): 57- 63.
[11]GIBSON D G, BENDERS G A, Andrews-Pfannkoch C, et al. Complete chemical synthesis, assembly, and cloning of aMycoplasmagenitaliumgenome [J]. Science, 2008, 319(5867): 1215-1220.
[12]GIBSON D G, GLASS J I, LARTIGUE C, et al. Creation of a bacterial cell controlled by a chemically synthesized genome [J]. Science, 2010, 329(5987): 52-56.
[13]GIBSON D G. Gene and genome construction in yeast [J]. Curr Protoc Mol Biol, 2011, 3(22): 1-17.
[14]GIBSON D G. Oligonucleotide assembly in yeast to produce synthetic DNA fragments [J]. Methods Mol Biol, 2012 (852): 11- 21.
【责任编辑李晓卉】
ApplicationofGibsonAssemblymethodforplantexpressionvectorconstruction
JI Zhicheng, JIANG Yanxiang, LIU Yaoguang, ZHANG Qunyu
(State Key Laboratory of Subtropical Agro-bioresources Conservation and Utilization/College of Life Sciences, South China Agricultural University, Guangzhou 510642, China)
【Objective】 To construct a convenient and efficient plant expression vectors. 【Method】 Gibson et al developed an isothermalinvitrorecombination system or Gibson Assembly for ligation of multiple DNA fragments. The primers were designed to amplify target DNA sequences that contained 20-25 bp overlapping ends. The target DNA segments,linearized vector and the enzyme mixture were mixed in one tube to perform the assembly reaction.【Result and conclusion】 Using this method, several vectors for expressing rice genes were assembled. To increase the efficiency for cloning multiple fragments, the assembled primary products by PCR were amplified and then ligated into the vector. The Gibson Assembly method is faster and simpler without the limitation of internal restriction endonuclease sites of target genes, and it can be applied widely to construction of various vectors.
Gibson Assembly; isothermalinvitrorecombination system; plant expression vector construction
2013- 06- 06优先出版时间2014- 07- 17
优先出版网址:http:∥www.cnki.net/kcms/detail/44.1110.S.20140717.0922.041.html
戢志呈(1989—),男,硕士研究生, E-mail: zhichengji@126.com;通信作者:张群宇(1973—),男,副研究员,博士,E-mail: zqy@scau.edu.cn
国家自然科学基金(30871331);973计划项目(2011CB100204)
戢志呈,江雁翔,刘耀光,等.应用Gibson Assembly方法构建植物表达载体[J].华南农业大学学报,2014,35(5):112- 116.
Q754
A
1001- 411X(2014)05- 0112- 05
猜你喜欢
环球时报(2022-09-20)2022-09-20 15:18:57
中学生数理化·中考版(2021年9期)2021-11-20 06:17:30
今日农业(2020年24期)2020-12-15 16:16:00
中等数学(2020年2期)2020-08-24 07:58:46
测控技术(2018年9期)2018-11-25 07:44:24
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:47:59
临床骨科杂志(2017年1期)2017-03-07 00:54:04
北京航空航天大学学报(2016年7期)2016-11-16 01:50:55
光学精密工程(2016年3期)2016-11-07 09:03:32
兽医导刊(2016年12期)2016-05-17 03:51:50
