
核磁共振检测食用植物油中有害多环芳烃蒽研究
2014-08-28 04:25吕梅香曾和平
华南师范大学学报(自然科学版) 2014年1期
吕梅香, 曾和平
(华南师范大学化学与环境学院,广州 510006)
多环芳烃(PAHs)产生于化石燃料、木材、垃圾或其他有机物(如烟草、烧焦的肉等)的不完全燃烧,且在空气、水、土壤中广泛分布. 多环芳烃具有很好的脂溶性,植物油原料受到PAHs污染以及加工方式不同等原因会导致食用油中含PAHs. 长期食用含有PAHs 的食用油对健康产生潜在威胁(致畸、致癌和致基因突变),萘、蒽等16种多环芳烃受到世界卫生组织的通缉,因此,探讨食用植物油中有害多环芳烃意义重大.
目前,食用油中PAHs的现行检测方法有:《GB/T 5009.27—2003食品中苯并(a)芘的测定》[1]规定的2种方法:提取分离、浓缩后的荧光分光光度法和目测比色法; 《GB/T 5009.22—2003食品中黄曲霉毒素B1的测定》[2]规定的提取、浓缩、薄层分离后在365 nm处的荧光强度法和提取、脱脂、浓缩后酶联免疫分析法.现行方法对于食用油中可能含有的其他多环芳烃检测标准尚未发布.
关于食用油中多环芳烃更快速、简便、准确的检测方法的研究主要有:HPLC-FL法[3]、GC-MS法[4]、恒能量同步荧光光谱法[5]等.上述方法大多需要对植物油先预处理(提取、富集、纯化)再检测的步骤来分析,具有步骤多、时间长、原料用量大、所需设备种类多、检测成本高等缺点,最大的不足是对样品的预处理可能使被测样品受损.
高分辨核磁共振法无需对样品进行任何处理,是一种对被测样品无损、快速、环保、不受提取分离过程中不确定因素干扰的方法. 早在上世纪80年代[6]便已用于分析植物油的主要成分,后来越来越多的学者用高分辨核磁共振法研究各种植物油的特征以及植物油的掺伪[7-16],分析植物油的主要成分、含量及峰形的细微变化,国内也有学者把高分辨核磁共振法用于研究地沟油,对区分各种油有较高的准确率[17-18].
核磁共振氢谱法能直观的告诉我们植物油中所含的不同类型的氢,如果植物油中含有多环芳烃,理论上也能够测出来,但目前还没有采用高分辨核磁共振法对植物油中多环芳烃的研究报道. 因此本文对4种常见植物油(棕榈油palm oil、花生油peanut oil、玉米油corn oil、橄榄油olive oil)中外源添加不同浓度的多环芳烃-蒽进行检测,为高分辨核磁共振法鉴别食用油中多环芳烃的最低限量做了探讨.
1 实验部分
1.1 试剂和仪器
试剂:棕榈油(鲜榨)、花生油(鲁花5S压榨1级浓香花生油,华润万佳超市)、玉米油(福临门,黄金产地,华润万佳超市)、橄榄油(刀唛特级初榨橄榄油,西班牙进口原料,华润万佳超市)、多环芳烃(蒽,分析标准品,>99.7%,阿拉丁试剂公司)、氘代氯仿(CDCl3,美国剑桥CIL公司).仪器:美国安捷伦公司VNMR SYSTEM 400 M液体核磁共振波谱仪,ATB探头,5 mm核磁管.
1.2 样品制备
1.2.1 纯植物油-CDCl3的配制 用10 mL CDCl3溶解50 g植物油,称取1 g置于核磁管中. 用于测试纯植物油的1H NMR;
1.2.2 蒽标准品-CDCl3的配制 准确称取1 mg蒽标准品用1 mL CDCl3溶解,置于核磁管中. 用于测试纯蒽的1H NMR;
1.2.3 含蒽植物油-CDCl3的配制 准确称取100 mg蒽标准品掺到100 g植物油中,使蒽充分溶于植物油后,加入10 mL CDCl3,制成蒽在植物油中含量为1 g/kg的植物油-CDCl3样品.
取含蒽1 g/kg的植物油-CDCl3样品1 g,加入1.2.1中的植物油-CDCl39 g,混匀后即制成蒽在植物油中含量为100 mg/kg的植物油-CDCl3样品; 同法逐级稀释配制含蒽10 mg/kg、1 mg/kg、100 μg/kg、10 μg/kg的植物油-CDCl3样品. 分别称取1 g置于核磁管中. 用于测试含蒽植物油的1H NMR.
1.3 测试条件
使用反式ATB探头测试1H NMR. 测试温度T为40 ℃,弛豫时间为d1为1 s,采样时间at为2 s,预采次数ss为 2,扫描次数nt为128.
2 结果与讨论
2.1 植物油的1H-NMR
植物油的主要成分是脂肪酸甘油三酯,即硬脂酸、软脂酸、油酸、亚油酸的甘油酯,花生油、玉米油、橄榄油、棕榈油也不例外. 以棕榈油的核磁共振氢谱1H NMR (以CDCl3定标,化学位移δ为7.24 ppm)为例(图1).
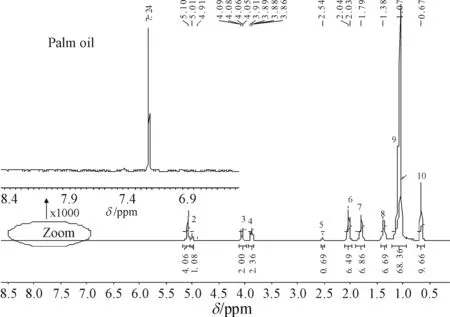
图1 棕榈油的核磁共振氢谱1H NMR
棕榈油的1H NMR位移值集中在0.00~5.50 ppm之间,图1中插图所示为δ=6.50~8.50 ppm之间的纵坐标放大1 000倍的谱图,可以看到除了溶剂峰外没有其他峰. 这与各种食用植物油1H NMR的主要成分谱图解析结果[7-8,10,13-15]一致,只是化学位移略有偏差,各特征峰积分面积不同. 4种植物油中,每种植物油每组特征峰的位移值都相近,峰面积明显不同(表1),分别代表了各种植物油中油酸、亚油酸、饱和脂肪酸甘油酯等的含量.
2.2 蒽标准品的1H-NMR
轻质多环芳烃-蒽的结构简单(<4个环),只有10个H原子. 在高分辨核磁共振仪中,由于质子受各向异性效应的影响使其产生的化学位移均在低场,且每组质子受该效应的影响不同使其化学位移不同.蒽处于相同化学环境的质子可分为3组,每组质子受相邻质子的影响产生耦合裂分.因此,在1H NMR图谱中更容易跟踪识别.蒽标准品在CDCl3中的1H NMR谱图如图2所示. 蒽的耦合常数见表2,从低场到高场的峰依次编号为A、B、C.
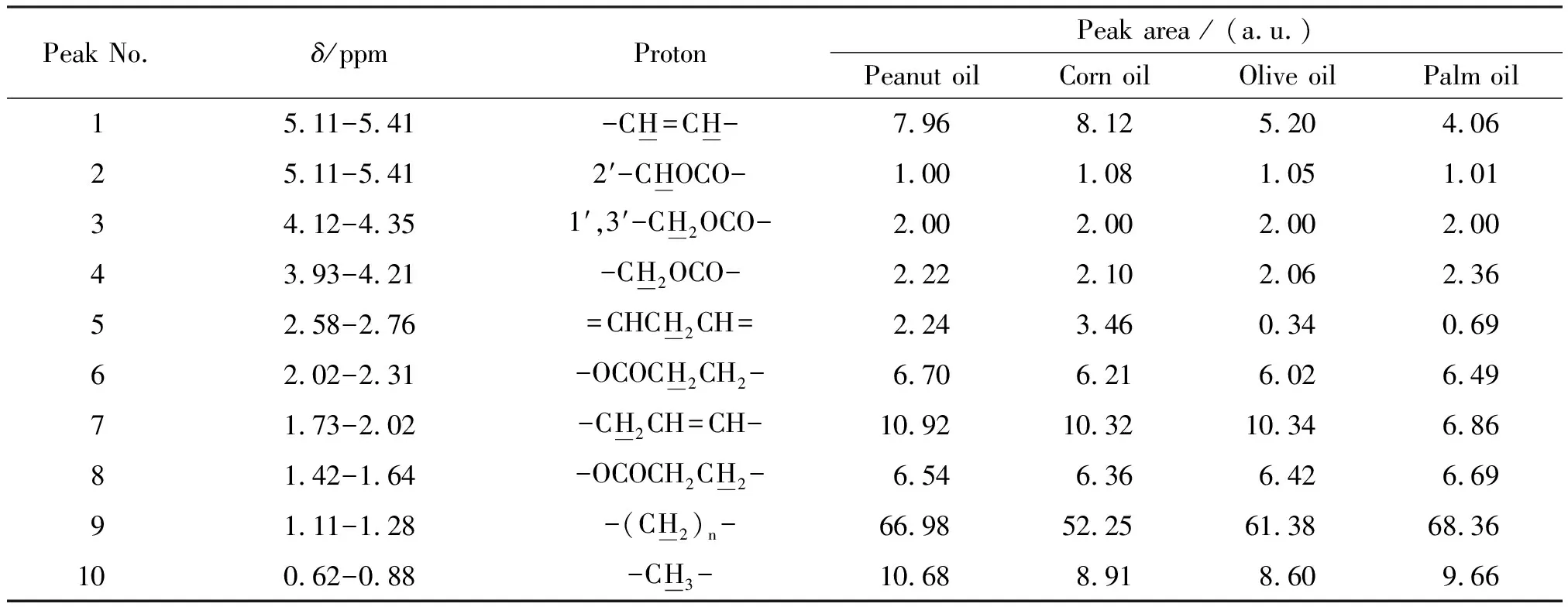
表1 各种植物油的化学位移和峰面积Table 1 The chemical shift and peak area of each group peak of vegetable oils

表2 纯蒽和掺入棕榈油后的蒽化学位移及耦合常数Table 2 The chemical shifts and coupling constants of anthracene in different vegetable oils

图2 蒽在CDCl3中的的核磁共振氢谱1H NMR
2.3 蒽掺入植物油后的1H NMR
植物油和蒽的1H NMR特征峰化学位移相差较远,植物油集中在δ=0.700~5.500 ppm之间,而蒽集中δ=7.400~8.500 ppm之间,且只有3组峰,分别为单峰(δ=8.410 ppm)及2个dd峰(δ=7.990 ppm和δ=7.440 ppm). 即植物油和蒽2种物质混合后的1H NMR峰理论上不会重叠.
实际上掺入植物油中的蒽量极少,其化学环境会受植物油的影响而发生变化,而油的量大,其化学环境几乎不受蒽的影响. 从图3看出,正常情况下,由于人们习惯观察的是样品的主要成分,不会把纵坐标无限放大来观察有无杂质,因此只能看到植物油的10组δ=0.000~5.500 ppm的特征峰,看不到其他峰. 当逐渐放大δ=6.500~8.500 ppm部分的纵坐标,蒽的特征峰逐渐清晰,图3的插图为纵坐标放大200倍的情况. 而且由于蒽的特征性,较容易识别. 不同含蒽量的植物油的1H NMR在δ=7.000~9.000 ppm之间峰高放大数倍(以蒽的峰能够识别,易与基线噪音区别为限)后都能看到化学位移、耦合常数未发生变化的3组特征峰.
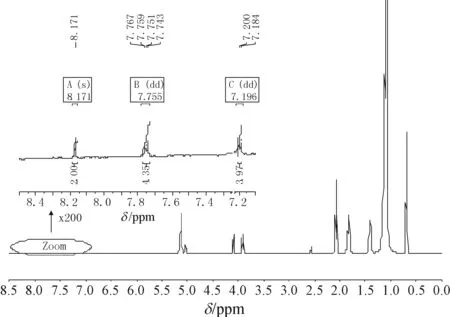
图3 含100 mg/kg蒽的棕榈油的核磁共振氢谱1H NMR
植物油中蒽的3组峰在棕榈油中向高场位移0.240 ppm,在橄榄油中向高场位移0.200 ppm,在玉米油中向高场位移0.150 ppm,在花生油向高场位移0.140 ppm. 蒽在植物油中的浓度在100 mg/kg至10 μg/kg之间的1H NMR峰形、峰面积、位移值、耦合常数完全一致,且与植物油的特征峰不重叠,容易区分. 表2为蒽纯品及掺入到植物油后的蒽表现出来的峰信息.
图4为含10 μg/kg蒽的棕榈油在δ=7.0~9.0 ppm范围的出峰情况,当纵坐标放大1 000倍时,有3组峰出现,且与表2中所列出的较高浓度蒽含量的化学位移、峰面积、耦合常数基本一致(误差原因是浓度极稀后,1H NMR的纵坐标放大达1 000倍,使得基线漂移,峰形不完全左右对称导致),因此可确定是蒽的1H NMR峰,此浓度已达到《食用植物油卫生标准GB 2716—2005》[19]规定每千克植物油含多环芳烃10 μg的检出限.
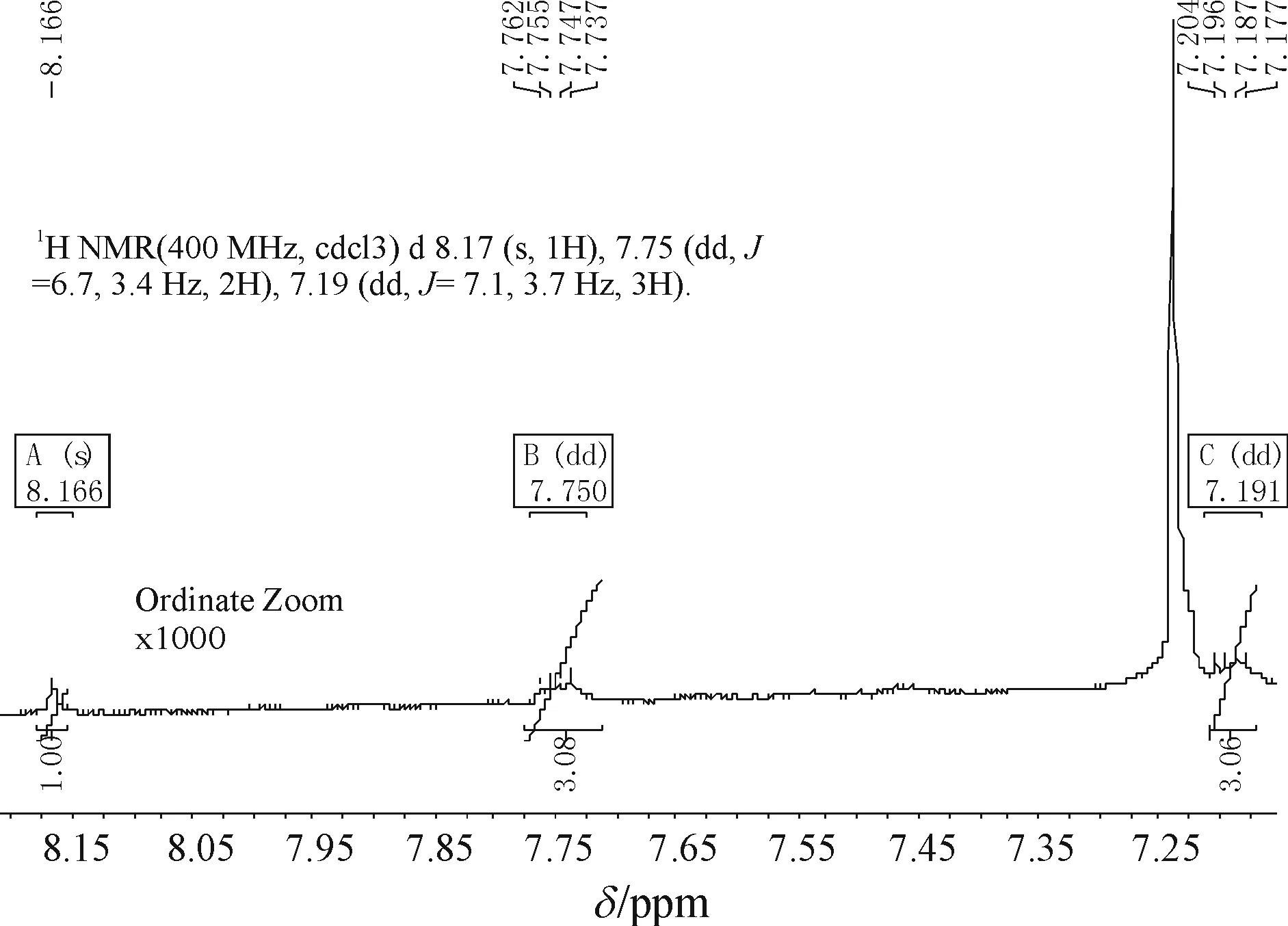
图4 含10 μg/kg蒽的棕榈油的核磁共振氢谱1H NMR
Figure 4 The1H NMR of palm oil containing 10 μg/kg anthracene
3 结论
中国国家标准《GB 2716—2005食用植物油卫生标准》规定,苯并(a)芘含量不得超过 10 μg/kg. 德国油脂科学学会(DGF)对油中重质 PAHs 推荐控制值为5 μg/kg,而对油中 PAHs 总量推荐控制值为 25 μg/kg; 欧盟法规(EC)No 208/2005规定限量为 2.0 μg/kg.
对食用油多环芳烃检测的国家标准中,只有苯并芘和黄曲霉素的检测方法,长期以来油的出厂报告中也没有其他10余种多环芳烃的检测报告.
本文采用在植物油中添加蒽的方式,用400 M核磁共振仪测试不同蒽含量的1H NMR谱图.蒽在植物油中,与纯蒽一样只有3组峰,1组单峰,2组dd峰,且远离植物油的特征峰组,容易识别,虽含量低,但是能观察到.
蒽在植物油中含量为10 μg/kg时仍能够分辨出,达到了国家标准对苯并芘的检测限要求.因此,核磁共振法法作为检测植物油中多环芳烃蒽存在与否的方法有效且对被测样品无损、快速、准确.
参考文献:
[1] 中华人民共和国国家标准. 食品中苯并(a)芘的测定[S]. GB/T5009.27—2003. 北京:中国标准出版社, 2004.
[2] 中华人民共和国国家标准. 食品中黄曲霉毒素B1的测定[S]. GB/T5009.22—2003. 北京:中国标准出版社, 2004.
[3] Lage Y M A, Cortizo D J L. Supercritical fluid extraction and high-performance liquid chromatography-fluorescence detection method for polycyclic aromatic hydrocarbons investigation in vegetable oil[J]. Food Control, 2005, 16 (1):59-64.
[4] Purcaro G, Morrison P, Moret S, et al. Determination of polycyclic aromatic hydrocarbons in vegetable oils using solid-phase microextraction-comprehensive two-dimensional gas chromatography coupled with time-of-flight mass spectrometry[J].Journal of Chromatography A, 2007, 1161(1/2):284-291.
[5] 邱如斌, 章汝平, 林水东, 等. 恒能量同步荧光光谱法测定食用油中多环芳烃[J]. 光谱实验室, 2011, 28(2):777-781.
Qiu R B, Hang R P, Lin S D, et al.Determination of Polycyclic Aromatic Hydrocarbons in edible oils by Constant-Energy Synchronous Fluorescence Spectrometry[J]. Chinese Journal of Spectroscopy Laboratory, 2011, 28(2):777-781.
[6] Kvalheim O M, Aksnes D W, Brekke T, et al. Crude oil characterization and correlation by principal component analysis of Carbon-13 Nuclear Magnetic Resonance Spectra[J]. Analytical Chemistry, 1985, 57(14):2858-2864.
[7] Guillén M D, Ruiz A, Cabo N. Characterization of Sacha inchi oil by FTIR Spectroscopy and1H NMR comparison with Linseed Oil[J]. Journal of the American Oil Chemists’ Society, 2003, 80(8):755-762.
[8] Miyake Y, Yokomizo K, Matsuzaki N. Determination of Unsaturated Fatty Acid composition by High-Resolution Nuclear Magnetic Resonance Spectroscopy[J]. Journal of American Oil Chemists’ Society, 1998, 75(9):1091-1094.
[9] Vlahov G, Giuliani A A, Del R P.13C NMR spectroscopy for determining the acylglycerol positional composition of lampante olive oils chemical shift assignments and their dependence on sample concentration[J]. Royal Society of Chemistry, 2010(2):916-923.
[10] Kumar R, Bansal V, Tiwari A K, et al. Estimation of glycerides and free fatty acid in oils extracted from various seeds from the Indian region by NMR spectroscopy[J]. Journal of American Oil Chemists’ Society, 2011, 88:1675-1685.
[11] Cordella C B Y, Tekye T, Rutledge D, et al. A multiway chemometric and kinetic study for evaluating the thermal stability of edible oils by1H NMR analysis: Comparison of methods[J]. Talanta, 2012, 88:358-368.
[12] Gunstone F D. The composition of hydrogenated fats by high-resolution13C Nuclear Magnetic Resonance Spectroscopy[J]. Journal of American Oil Chemists’ Society, 1993, 70(10):965-970.
[13] Falch E, Anthonsen H W, Axelson D E, et al. Correlation between1H NMR and traditional methods for determining lipid oxidation of ethyl docosahexaenoate[J]. Journal of American Oil Chemists’ Society, 2004, 81(12):1105-1110.
[14] Vigli G, Philippidis A, Spyros A, et al. Classification of edible oils by employing31P and1H NMR spectroscopy in combination with multivariate statistical analysis:A proposal for the detection of seed oil adulteration in virgin olive oils[J]. Journal of Agriculture and Food Chemistry, 2003, 51:5715-5722.
[15] Hidalgo F J, Zamora R. Edible oil analysis by high-resolution nuclear magnetic resonance spectroscopy: Recent advances and future perspectives[J]. Trends in Food Science & Technology, 2003, 14:499-506.
[16] Hoffman D R, Collins-Williams C. Cold-pressed peanut oil may contain peanut allergens[J]. Journal of Allergy and Clinical Immunology, 1994, 93:801-802.
[17] 许秀丽, 任荷玲, 李娜, 等. 核磁共振技术在地沟油鉴别中的应用研究[J]. 检验检疫学刊, 2012, 22(4):25-32.
Xu X L, Ren H L, Li N, et al. Application of Nuclear Magnetic Resonance technology in hogwash oil determination[J]. Inspection and Quarantine Science, 2012, 22(4):25-32.
[18] 蔡波太, 袁龙飞, 周影, 等. 基于1H NMR指纹图谱结合多变量分析的地沟油检测方法[J]. 中国科学:化学, 2013, 43(1):1-10.
Cai B T, Yuan L F, Zhou Y, et al. Identification of illegal cooking oils by using1H NMR fingerprints combined with multivariate analysis[J]. Scientia Sinica: Chimica, 2013, 43(1):1-10.
[19] 中华人民共和国国家标准. 食用植物油卫生标准[S]. GB2716—2005, 北京:中国标准出版社, 2005.
猜你喜欢
制造技术与机床(2022年5期)2023-01-06
油气藏评价与开发(2022年2期)2022-11-27
现代临床医学(2022年4期)2022-09-29
现代食品科技(2022年8期)2022-09-02
中国抗生素杂志(2022年7期)2022-08-18
长春师范大学学报(2019年4期)2019-04-29
中国油脂(2019年1期)2019-01-23
世界热带农业信息(2019年12期)2019-01-05
世界热带农业信息(2018年6期)2018-03-05
世界热带农业信息(2018年7期)2018-01-19
