
胰岛素及格列齐特治疗2型糖尿病大鼠肝脏脂质沉积的机制探讨*
2014-08-08 09:23:30刘宏霞严晋华翁建平
中国病理生理杂志 2014年6期
袁 丁, 梁 华, 刘宏霞, 许 婧, 徐 芬, 严晋华, 翁建平
(中山大学附属第三医院内分泌与代谢病科,广东 广州 510630)
2型糖尿病以胰岛素抵抗为特征,而肝脏是胰岛素作用的靶器官之一,脂肪在肝脏的异位沉积也是导致外周组织胰岛素抵抗的一个重要因素。课题组前期研究发现,新诊断的2型糖尿病患者分别经胰岛素强化治疗和口服药物强化治疗后,均能改善β细胞功能及胰岛素敏感性,但2种治疗方案的血糖控制达标率、达标时间和1年后缓解率不同[1]。这种差异与肝脏脂质沉积改善的分子机制是否相关尚不明确。在高脂饮食和小剂量链脲佐菌素(streptozocin,STZ)诱导的SD大鼠2型糖尿病模型当中,早期胰岛素和格列齐特治疗均能改善糖尿病大鼠肝脏胰岛素抵抗[2],因而我们设想两者改善大鼠肝脏胰岛素抵抗的分子机制可能不尽相同。因此,我们利用胰岛素及格列齐特治疗高脂饮食和STZ诱导的2型糖尿病大鼠模型,观察其对肝脏脂质沉积、脂质代谢通路及内质网稳态的影响并探讨2种药物治疗的分子机制。
材 料 和 方 法
1 动物模型建立及干预
7~8周龄雄性SD大鼠,体重180~200 g,适应性喂养2周后,根据体重随机分组,给予普通饲料(脂肪10%)和高脂饲料(脂肪60%,Research Diets)喂养5周。高脂饮食组大鼠腹腔注射1%链脲佐菌素(35 mg/kg; Sigma)。链脲佐菌素注射后第3天,取尾静脉血测空腹血糖,空腹血糖>13.8 mmol/L为糖尿病大鼠。将成模的糖尿病大鼠随机分为:(1) 糖尿病组(diabetes mellitus,DM;n=10);(2) 早期胰岛素治疗组(insulin,INS;低精蛋白锌人胰岛素,6~8 U/d,皮下注射;n=10);(3)格列齐特治疗组(gliclazide per os,PO;80 mg·kg-1·d-1,灌胃;n=10)。治疗3周结束后3 d处死大鼠,取全血或血清检测生化指标,取肝脏组织称量后置液氮,后转移至-80 ℃冰箱保存。
2 相关指标检测
空腹血糖用雅培Optium血糖试纸测定,血清甘油三酯(triglyceride, TG)和总胆固醇(total choleste-rol, TC)采用酶法测定。
3 肝脏组织油红O染色
制作大鼠肝脏组织冰冻切片(厚度0.5 μm),70%乙醇固定1 min后用蒸馏水漂洗,加入油红稀释染液避光密封染色15 min,放入60%乙醇中镜下分化至间质清晰后用蒸馏水漂洗,再加入Marry苏木素染核1 min,蒸馏水漂洗后明胶封片,正置显微镜(Leica)观察并摄片,脂肪小滴呈红色,细胞核呈蓝色,间质无色。
4 血清脂联素(adiponectin)水平检测
参照大鼠脂联素酶联免疫试剂盒(RayBio)说明书,将与辣根过氧化物酶结合的抗生物素蛋白加入96孔板中,加入标准品及受检大鼠血清样本进行孵育,与四甲基联苯胺(tetramethylbenzidine,TMB)底物溶液显色后加入硫酸终止反应,用酶标仪在450 nm处检测。绘制标准曲线得出样本的脂联素浓度。
5 Western blotting
冰浴中匀浆肝脏组织,提取总蛋白(RIPA Lysis and Extraction Buffer,PIERCE)、核蛋白(Merck),用BCA法测定蛋白浓度,根据蛋白分子量大小分别用7%,10% SDS-PAGE,转至PVDF膜(Whatman)上。室温下用5%脱脂奶粉封闭2 h,然后与相应抗体AMPK(AMP-activated protein kinase)、Thr172p-AMPK (AMPK phosphorylated on threonine 172)、Ser372p-SREBP-1c (SREBP-1c phosphorylated on serine 372)、 ACC (acetyl-CoA carboxylase)、 Ser79p-ACC (ACC phosphorylated on serine79)、BiP (immunoglobulin-binding protein)和β-actin(CST)。SREBP-1c (sterol regulatory element-binding protein 1c)抗体(Abcam)4 ℃孵育过夜,次日以TBST洗膜,再用相应荧光Ⅱ抗IRDye 800CW Secondary Antibodies(LI-COR Biosciences)室温避光孵育1h,洗膜后用Odessey仪器(LI-COR Biosciences)曝光显影。
6 实时荧光定量PCR
提取大鼠肝脏组织总RNA(Trizol,Invitrogen),将1 μg总RNA逆转录为cDNA(PrimerScript RT reagent Kit,TaKaRa)。使用SYBR Premix Ex Taq(TaKaRa)试剂盒进行扩增,反应体系:cDNA 2 μL,上、下游引物(序列见表1)各0.5 μL,SYBR buffer 10 μL,去离子水7 μL。反应条件:95 ℃预变性 30 s;95 ℃ 5 s,60 ℃ 20 s,40个循环。用Ct法(2-ΔΔCt)比较各mRNA表达水平。
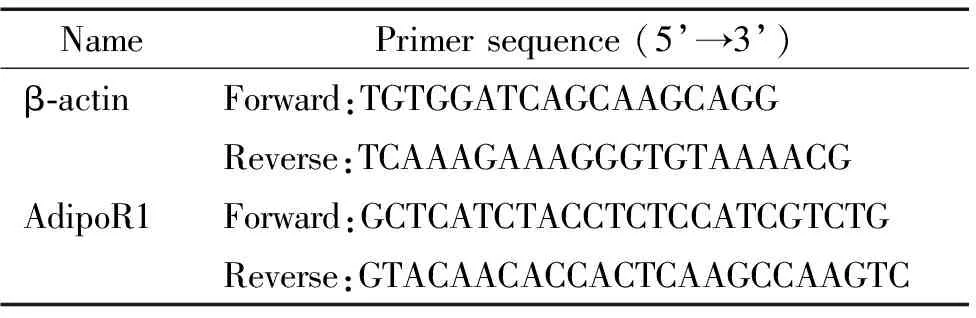
表1 引物序列
7 统计学处理
采用SPSS 17.0软件分析,计量资料以均数±标准差(mean±SD)表示,多组资料比较采用方差分析,两两比较采用最小显著差异(LSD)法。以P<0.05(双侧)为差异有统计学意义。
结 果
1 各组大鼠的代谢及生化学指标
DM大鼠体重无明显变化,空腹血糖及血甘油三酯显著升高。经胰岛素及格列齐特治疗后,体重有明显增加,空腹血糖得到改善(P<0.05),但仍高于NC组(P<0.05),TG降至NC水平。TC各组间的差异均无统计学意义(均P>0.05),见表2。

表2 SD大鼠生化及代谢指标
2 肝脏脂质沉积情况
DM大鼠肝脏脂滴较NC组明显增多,胰岛素及格列齐特治疗后肝脏脂质沉积得到改善,且INS组更为明显,见图1。
3 治疗对血清Adiponectin及肝脏AdipoR1 mRNA的影响
DM组血清Adiponectin及肝脏AdipoR1 mRNA水平较NC组明显降低(P<0.05),分别下降52.0%和48.2%。胰岛素治疗后均明显升高(P<0.05),血清adiponectin较DM组升高了5.3倍,肝脏AdipoR1 mRNA 较DM组升高4.4倍。格列齐特治疗后血清adiponectin及肝脏AdipoR1 mRNA水平恢复至NC组水平(均P<0.01),见图2。
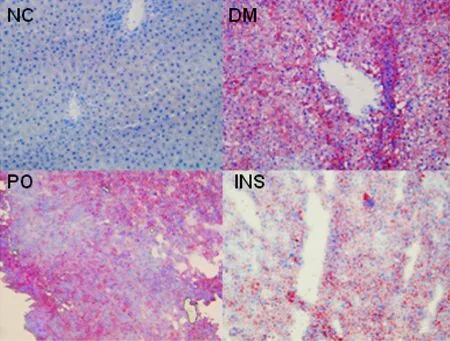
Figure 1. The hepatic oil red O staining in rats (×200) . NC: normal control; DM: diabetes mellitus; INS: diabetic rats treated with insulin; PO: diabetic rats treated with gliclazide per os.
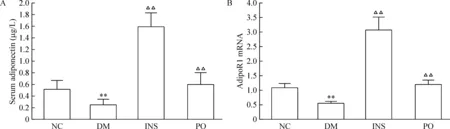
Figure 2. The levels of serum adiponectin (A) and AdipoR1 mRNA expression (B) in liver.NC:normal control; DM: diabetes mellitus; INS: diabetic rats treated with insulin; PO: diabetic rats treated with gliclazide per os.Mean±SD.n=6.**P<0.01 vs NC; △△P<0.01 vs DM.
4 治疗对Thr172p-AMPK/AMPK表达的影响
DM大鼠AMPK与Thr172p-AMPK均较NC组减少,Thr172p-AMPK/AMPK表达比值(0.25±0.04)较NC(0.67±0.05)下降62.5%(P<0.01)。格列齐特治疗后Thr172p-AMPK/AMPK恢复至NC组水平(0.63±0.01),主要体现在Thr172p-AMPK表达量增加,而总AMPK较DM组无明显变化。胰岛素治疗后AMPK与Thr172p-AMPK的表达量均增加,且Thr172p-AMPK的表达较DM组增加显著,Thr172P-AMPK/AMPK的表达比值(1.23±0.08)较DM组增加了3.87倍,较NC组增加了0.83倍(P<0.01),见图3。
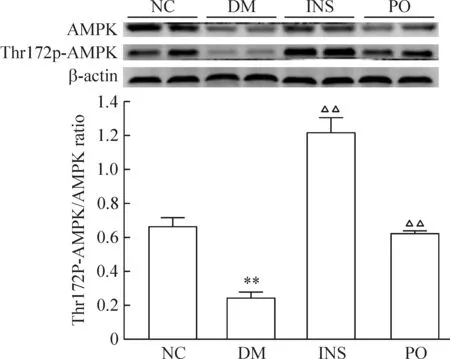
Figure 3. Thr172p-AMPK/AMPK protein expression in liver.Mean±SD.n=6.**P<0.01 vs NC; △△P<0.01 vs DM.
5 治疗对Ser372p-SREBP-1c/SREBP-1c表达比值和BiP的影响
与NC组(0.90±0.08)比较,DM大鼠SREBP-1c表达明显升高,Ser372p-SREBP-1c 水平和Ser372p-SREBP-1c/SREBP-1c比值明显下降(0.22±0.06),下降幅度为75.2%(P<0.01)。胰岛素治疗后,SREBP-1c表达显著降低,Ser372p-SREBP-1c水平明显升高,Ser372p-SREBP-1c/SREBP-1c(1.35±0.09)比值较DM组增加5.01倍,较NC组增加了0.49倍(P<0.01)。格列齐特治疗后,SREBP-1c表达与DM组相比下降(P<0.01),但ser372p-SREBP-1c水平明显升高,其比值恢复至NC组水平(0.90±0.04)。胰岛素组SREBP-1c表达水平与格列齐特组相比有显著差异(P<0.05),两治疗组Ser372p-SREBP-1c/SREBP-1c差异也有统计学意义(P<0.05),见图4A。与NC组(0.42±0.01)比较,DM大鼠BiP蛋白表达明显增加(0.69±0.01,P<0.01),胰岛素和格列齐特治疗后,BiP表达均有所减少,胰岛素组BiP表达下降42.9%(P<0.01),恢复至NC组水平,格列齐特组下降65.6% (P<0.01),降到NC组水平以下,见图4B。
6 治疗对Ser79p-ACC/ACC表达比值的影响
DM大鼠Ser79p-ACC/ACC表达比值为(0.45±0.02),较NC组(0.95±0.03)降幅为52.6%(P<0.01),主要体现在Ser79p-ACC明显减少。胰岛素治疗后其比值恢复至NC组水平(0.94±0.02),而格列齐特治疗后无明显变化(P<0.01),见图5。
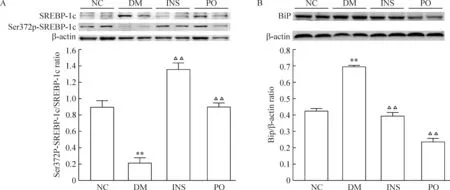
Figure 4. Ser372p-SREBP-1c/SREBP-1c (A) and BiP (B) protein expression in liver.Mean±SD.n=6.**P<0.01 vs NC; △△P<0.01 vs DM.
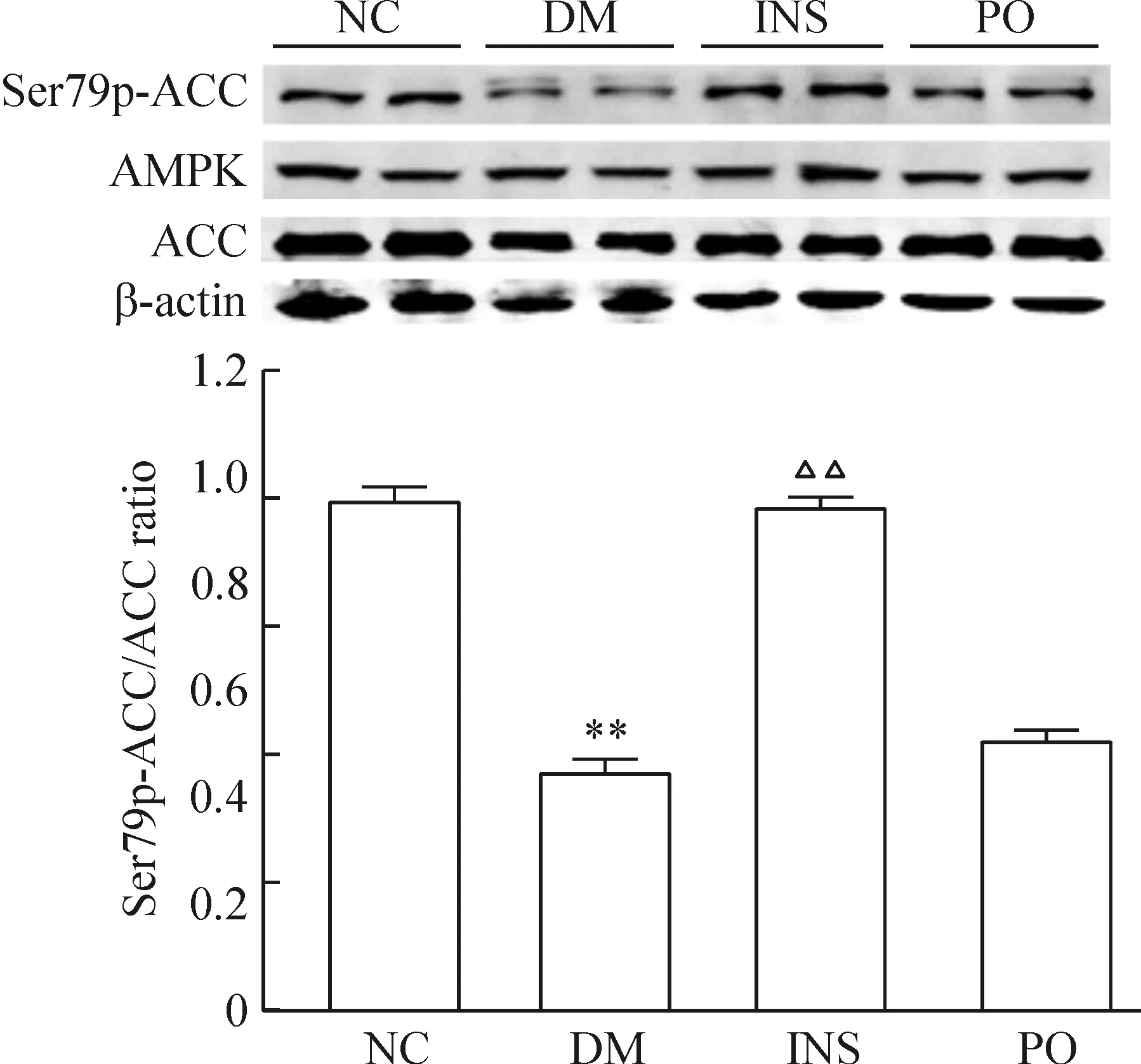
Figure 5. Ser79p-ACC/ACC expression in liver.Mean±SD.n=6.**P<0.01 vs NC; △△P<0.01 vs DM.
讨 论
肝脏是胰岛素作用的重要靶器官。肝脏胰岛素敏感性下降将促发肝脏脂质合成,增加甘油三酯含量。我们前期研究显示短期胰岛素或格列齐特强化治疗均能改善肝脏胰岛素抵抗,但具体分子机制尚不明确[2]。肝脏胰岛素抵抗与肝脏过度脂肪堆积密切相关。脂肪酸氧化与合成在维持肝脏脂质平衡中发挥重要的作用。
脂联素是一种脂肪组织特异性分泌的脂肪因子,与胰岛素抵抗呈负相关关系,可以通过结合其受体(AdipoR1)激活AMPK从而影响细胞内脂质代谢过程[3]。AMPK是调控肝脏脂质代谢的核心因子,既能通过抑制SREBP-1c的作用减少脂肪酸合成,也能通过磷酸化ACC,从而解除malonyl-CoA对CPT-1的抑制作用增加脂肪酸氧化[4]。因此,AMPK是参与胰岛素或格列齐特改善肝脏胰岛素抵抗机制的关键因子。本研究中,糖尿病组血清脂联素水平及其受体adipoR1表达均明显降低,说明糖尿病组存在胰岛素抵抗,而胰岛素与格列齐特治疗后均能提高脂联素及adipoR1表达的水平,激活AMPK,改善胰岛素抵抗,此结果与他人报道的结果相似[5-6]。提示2种药物存在共同的作用机制,但两者对脂联素-AMPK的激活程度不尽相同,提示激活的AMPK的后续作用和效应,两者可能存在差异。
SREBP-1c为脂质合成调控的关键转录因子,AMPK对SREBP-1c的调控现已知有2条途径,即通过直接磷酸化SREBP-1c丝氨酸372位点抑制SREBP-1c入核[7]及抑制内质网应激间接抑制SREBP-1c表达[8]。在我们的研究中,外源性胰岛素激活AMPK既通过磷酸化对SREBP-1c产生短期影响,也通过抑制SREBP-1c的表达产生长期影响。而格列齐特刺激的内源性胰岛素对SREBP-1c仅产生磷酸化的短期影响,这可能是胰岛素较格列齐特更有利于长期控制血糖、改善胰岛素抵抗的机制之一。此外,我们研究发现,胰岛素改善肝脏脂肪沉积的机制还体现在其能通过磷酸化ACC促进脂肪酸氧化,而格列奇特则无此作用,这也是胰岛素强化治疗为何优于格列齐特的原因之一。AMPK尚能通过抑制内质网应激抑制SREBP-1c的激活和表达。既往研究发现胰岛素及格列齐特治疗均能改善糖尿病大鼠肝脏的内质网应激,我们的研究也有类似的发现,但格列齐特组与胰岛素组相比BiP的抑制更为明显,这一点与之前的研究不符[2]。其原因可能是本研究胰岛素组的血糖控制与格列齐特组相当,而之前的研究胰岛素组的血糖控制比格列齐特组更好所致。
肝脏脂质平衡的机制极其复杂,尽管我们有了一些发现,但是除了脂联素-AMPK通路之外,可能还存在其它机制。瘦素(leptin)也是其中一种脂肪因子,其作用可通过STAT3途径介导脂肪生成[9],也能通过JAK2依赖途径激活AMPK介导脂肪酸氧化[10]。此外,脂联素也能通过AdipoR2激活PPARα介导脂肪酸氧化[11]。因此,胰岛素和格列齐特改善肝脏脂肪沉积、改善胰岛素抵抗的作用机制还有待更多研究去发现。
综上所述,我们发现胰岛素治疗和格列齐特口服药治疗均能通过激活脂联素-AMPK减轻2型糖尿病大鼠肝脏的脂质沉积。但两者具体分子机制有所不同,胰岛素激活AMPK,通过抑制SREBP-1c表达、直接磷酸化SREBP-1c抑制SREBP-1c入核等短期和长期的作用以及抑制内质网应激影响SREBP-1c,减少脂质合成;此外,胰岛素还通过激活AMPK增加ACC磷酸化增加脂质氧化。而格列齐特仅通过磷酸化的短期作用和抑制内质网应激对SREBP-1c产生影响,且对脂肪酸氧化无作用。两者机制上的差异可能部分解释临床上胰岛素较格列齐特更有利于长期控制血糖、改善胰岛素抵抗。
[参 考 文 献]
[1] Weng J, Li Y, Xu W, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial[J]. Lancet, 2008,371(9626):1753-1760.
[2] 孙卫平,毕 艳,梁 华,等. 早期胰岛素治疗对糖尿病大鼠肝脏固醇调节级联反应和脂肪沉积的影响[J]. 中华医学杂志,2011,91(26):1809-1812.
[3] Combs TP, Marliss EB. Adiponectin signaling in the liver[J]. Rev Endocr Metab Disord, 2014,15(2):137-147.
[4] Steinberg GR, Kemp BE. AMPK in health and disease[J]. Physiol Rev, 2009,89(3):1025-1078.
[5] Xynogala I, Pepelassi E, Perrea D, et al. Adiponectin and interleukin-6 levels in insulin-treated diabetic rats with experimental periodontitis[J]. Braz Oral Res, 2012,26(1):71-76.
[6] Drzewoski J, Zurawska-Klis M. Effect of gliclazide modified release on adiponectin, interleukin-6, and tumor necrosis factor-alpha plasma levels in individuals with type 2 diabetes mellitus[J]. Curr Med Res Opin, 2006,22(10):1921-1926.
[7] Li Y, Xu S, Mihaylova MM, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice[J]. Cell Metab, 2011,13(4):376-388.
[8] Tiano JP, Mauvais-Jarvis F. Molecular mechanisms of estrogen receptors’ suppression of lipogenesis in pancreatic beta-cells[J]. Endocrinology, 2012,153(7):2997-3005.
[9] Machinal-Quelin F, Dieudonne MN, Leneveu MC, et al. Proadipogenic effect of leptin on rat preadipocytesinvitro: activation of MAPK and STAT3 signaling pathways[J]. Am J Physiol Cell Physiol, 2002,282(4):C853-C863.
[10] Uotani S, Abe T, Yamaguchi Y. Leptin activates AMP-activated protein kinase in hepatic cells via a JAK2-dependent pathway[J]. Biochem Biophys Res Commun, 2006,351(1):171-175.
[11] Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions[J]. Nat Med, 2007,13(3):332-339.
猜你喜欢
中老年保健(2021年5期)2021-08-24 07:06:20
小雪花·成长指南(2021年2期)2021-05-20 09:14:00
天津医科大学学报(2019年3期)2019-08-13 06:53:00
初中生世界·九年级(2019年4期)2019-05-05 01:07:12
中西医结合心血管病电子杂志(2019年1期)2019-02-18 01:26:56
糖尿病新世界(2018年16期)2018-12-21 11:19:14
特别健康·下半月(2018年4期)2018-07-02 08:27:28
糖尿病新世界(2017年14期)2017-12-05 13:23:49
胃肠病学(2016年7期)2016-03-13 23:11:56
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
