
南方某尾矿库浅层尾砂中铀分布特征
2014-07-21 07:07占凌之朱晓杰
有色金属(矿山部分) 2014年3期
占凌之,朱晓杰,高 柏,杜 洋,汪 勇
(1.东华理工大学,江西 抚州 344000;2.赤峰九桦化工有限责任公司,内蒙古 赤峰 024000)
铀尾矿、废石中含有的放射性核素具有种类多、寿命长的特点,其中铀、镭、钍等核素所产生的迁移扩散已经对环境构成长久的潜在性危害。由《核工业三十年辐射环境质量评价》可知,铀矿冶地域通过水气途径对环境、居民最大有效剂量达到0.65~1.67mSv/a,集体有效剂量达到 17.57 人·Sv/a,而铀矿冶系统对居民集体剂量贡献占整个核燃料循环系统总集体贡献的93%,铀尾矿、废石对环境的剂量贡献占铀矿冶的80%[1]。铀是典型的放射性重金属元素之一,其毒性很强,一般表现为化学毒性和放射性毒性这两个方面[2-4]。铀进入人体的方式主要有以下几种:通过呼吸道吸入含铀的气溶胶、饮用被铀污染的水或者通过食物链进入体内[5]。因为铀及其子体均会在衰变过程中产生大量的α射线和β射线,所以会导致患骨癌和肺癌的风险增加[6-7]。尾矿砂虽然是铀水冶的废弃物,但还有5%铀、95%镭、75%钍等放射性核素存在尾矿砂中[8],尾矿砂在尾矿库中沉淀下来,产生的废水有可能通过各种途径或渗透到地下水或渗滤出尾矿库附近的地表水甚至直接排到农田,给当地居民和生态环境带来长久性危害。本文选择该尾矿库中铀分布特征及其形成机理进行研究,探讨尾矿库对矿区的环境和居民健康的影响,具有重要意义。
1 材料与方法
1.1 样品采集及制备
采样工具:铁锹、小铲、米尺、取样袋等。
选择尾矿库中的尾矿砂为本次调查对象,砂样采集于尾矿库浅层尾砂,尾矿库地势平坦。本文仅选取了面积为600m2左右的长方形区域进行布点采样,布点采用对角线布点法,共布4个点,每个样点相距约10m,分别在每个点处挖掘出一剖面(大于1m),在每个剖面上距地面0、20、40、60、80、100 cm深处的同一平面上集中一点分别采集一份砂样,共采集砂样24个,采集的每份砂样质量约1kg。样品用塑料袋密封,贴标签标明样号、深度、采集地点、日期。采样布点图见图1。

图1 尾矿砂样采样点分布图Fig.1 The distribution of sampling points of tailings sand
1.2 实验仪器和方法
1.2.1 实验设备
SHZ—82A型气浴恒温振荡器(金坛市荣华仪器制造有限公司);722N可见分光光度计(上海精密科学仪器有限公司);TDL—40B型高速离心机(上海安享科学仪器厂);DHG—9030A型加热恒温鼓风干燥箱(上海精宏实验设备有限公司);722型分光光度计(波长范围420~720nm)(上海精密仪器有限公司);分析天平(精度为0.0001g)(上海天平仪器厂);研钵;干燥器以及其他常规玻璃仪器。
1.2.2 实验方法
称取处理后的矿样10g置于250mL锥形瓶中,然后按照1∶10的固液比加入4%的硫酸100 mL,将三角锥形瓶垂直固定在振荡器上,调节振幅在(130±10)次/min,保持常温振荡。每隔2h进行一次离心,离心后取上清液1mL于10mL比色管中以备待测。
采用硫酸浸出尾矿砂样中的铀。在实验过程中,每2h就要对样品中的铀含量测定一次,每一次均是用移液管从每个锥形瓶中取出摇匀的浊液在离心机上分离,分离过后再把离心管中的液体再次摇成浊液倒回到锥形瓶中,以备连续测定。待测定结果稳定后,再进行离心,上清液经过测定后弃去,换等量的浸出液继续浸出实验,若测定结果再次达到稳定,则再一次换等量的浸出液,直至测不出铀浓度数值为止。
取制备好的待测液1mL于10mL比色管中,滴加两滴2,4-二硝基酚指示剂,用氨水(1+1)调至溶液呈黄色,然后用盐酸(1+3)调至溶液无色,再过量2滴,随即加入1mL缓冲溶液,1mL偶氮胂III溶液(0.05%),用去离子水稀释至刻度,摇匀,静置5min后,用3cm的比色皿,以空白试剂(去离子水)作参比,在660nm处测其吸光度并记录数据。
2 结果分析及讨论
2.1 各个尾矿砂中铀的含量及各动态变化
对不同距离、不同深度尾矿砂样连续浸出30h后,1、2、3、4号样浸出铀的含量见图2。
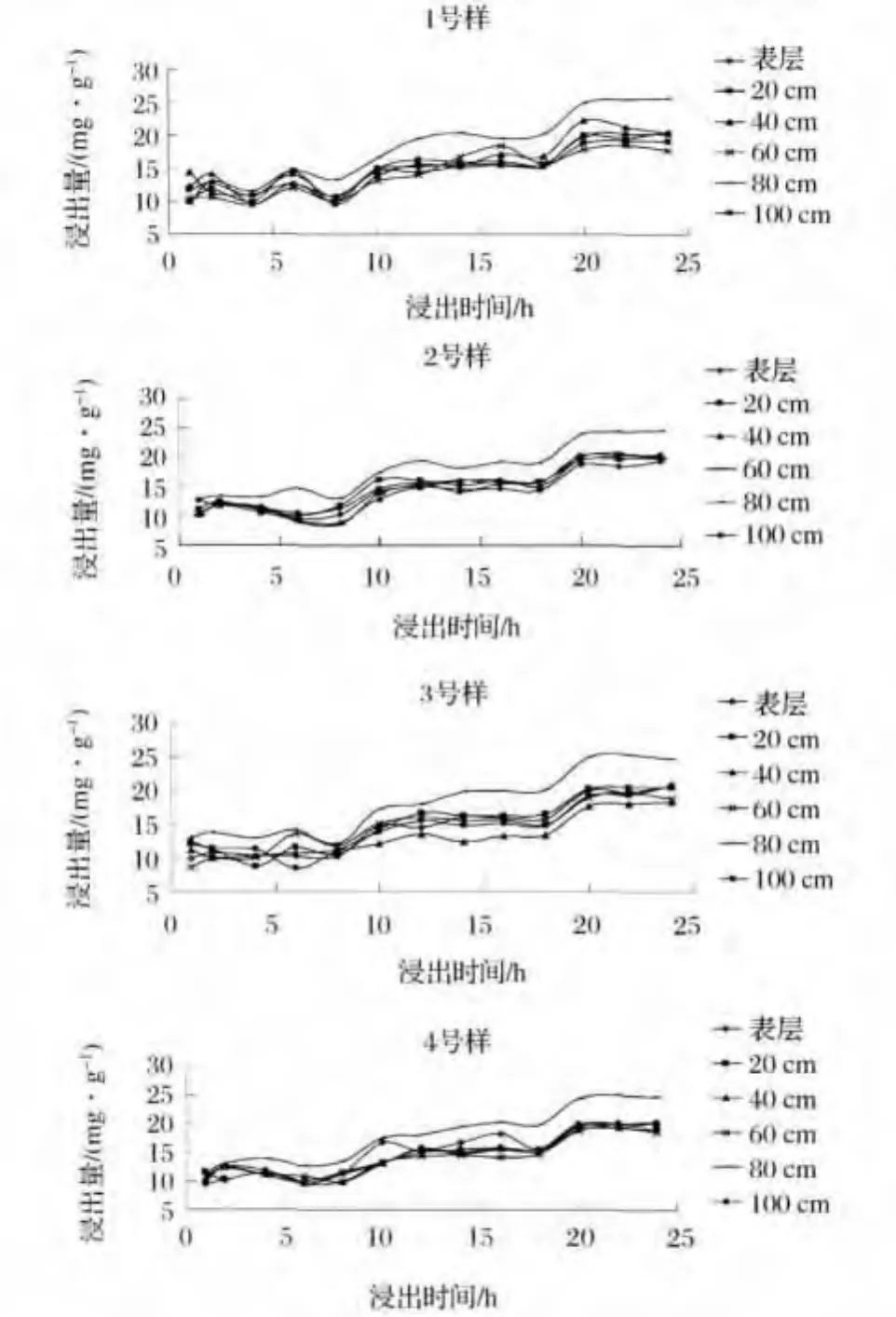
图2 尾矿砂样中铀浸出图Fig.2 Diagram of uranium leaching in the tailings sand
由图2可见,四个尾矿砂样铀的浸出结果规律有些差异。主要原因在于所取尾矿砂样处于不同的位置,降水、压实作用都不会完全相同,同时实验操作也会对所得数据有一定的影响。虽然在浸出时间10h之内浸出量并没有明显的变化,总体上铀的浸出量随着浸出时间的延长而增加。说明尾矿砂中各个层位中都含有一定量的铀存在,经计算四个样中0、20、40、60、80、100cm 最大值 分 别 为 20.69、20.51、20.33、19.81、25.63、20.34mg/g。
2.2 铀在尾矿砂中的垂直分布
由图3可见,尾矿砂中铀的含量分布并不是均匀的,也不与深度呈线性关系,但显然看出四个样中均为深度为80cm的尾矿砂样中铀的含量最高,1、2、3、4号砂样80cm的浸出量分别为25.63、24.57、24.84、24.86mg/g。由此可以得出,首先,在一定深度范围内,尾矿砂中铀是向下迁移的,在80cm深度达到最大。这可能是由于降雨以及尾矿库丰水期的综合影响。主要原因为:1)由于铀尾矿所处的地理位置可知,相山铀矿地处亚热带气候区,高温多雨,风化较强烈,且属于酸雨较严重的地域。实验测得铀尾矿砂显碱性,pH为7~10,经雨水淋滤后,会使在碱性条件下络合沉淀的铀溶解。若降雨量不大,雨水会慢慢从尾矿库表层下渗到深层尾矿砂处,矿砂中的铀也会随之下渗;若雨水量较大,雨水则会在尾矿库表层产生径流,表层部分矿砂会随径流流至较低处,则表层矿砂中的铀也会随之流走。所以无论雨量大小,雨水的淋滤作用都会对尾矿砂中的铀产生影响。吴晓燕等[9]研究表明降雨入渗率与入渗质量溶度对地下水核素溶度的影响关系。2)尾矿库丰水期,尾砂处于水面之下,处于一个厌氧环境,再加上水库三面环山,这个时期从山上随流水进入尾矿库及尾矿库本身的有机质体促进了此时尾矿砂中铀的向下迁移,Gustavo等[10]研究表明在厌氧条件下,存在的有机配体可能阻碍铀的沉淀物的形成及生物还原,从而促进铀在环境介质中的迁移。
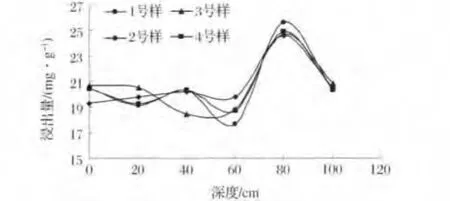
图3 尾矿砂样中铀浸出量Fig.3 The amount of uranium leaching in tailings sand
尾矿砂的粒度一般较细小,85%的粒度小于0.45mm,在经过石灰乳中和后,其尾矿质量分数一般在10%~25%,在长期的风化氧化作用下,粗砂及细泥极易离析。离析后的尾矿细泥堆密度更小,其沉积体渗透性也会更差。从取样现场看,表层至80cm,砂层疏松,愈深愈结实;80cm以下至100 cm,砂层紧密、结实。从表层至80cm深,尾矿砂大致可分为氧化环境、氧化-还原环境、还原环境。
从铀的浸出量看,除了尾矿砂中80cm铀的浸出量最大外,其它各个层位深度都不太高,保持低浓度状态。
铀在尾矿砂中的迁移受到多因素的影响,首先,降雨作用以及丰水期的促进作用,氧化还原环境会导致的铀溶解、沉淀。其次,胶体、温度梯度等因素也会导致铀在砂中的扩散,离子的吸附解吸。铀在氧化条件下容易发生迁移,而在还原条件下却几乎不迁移,所以介质(尾砂)的氧化还原环境对于铀的迁移起着重要作用。铀处于氧化环境中,利于水中U(VI)的迁移;反之,U(VI)则沉淀下来[11]。Dmckay等[11-12]认为地质介质的各向异性及胶体微粒的自身性质(如粒径、所带电荷的电性)对胶体的迁移速度和迁移距离有着至关重要的影响。曹存存等[13]研究发现在一定条件下胶体可以增强铀离子的迁移,而在另一种条件下胶体也可阻滞铀的迁移。此外铀在矿物中存在形式也对铀的迁移产生影响,铀以吸附态和铀矿物形式存在,易浸出的是吸附态以及与石英长石共生的铀矿物,而与黄铁矿共生并被石英包裹的铀矿物难以浸出[14]。因此,随着铀元素迁移的不断累积,尾矿库中尾矿砂表层的铀含量会小于其它各层。能溶解的铀基本被溶解迁移,随着深度的增加,下层矿砂受到的压实作用增强,易形成还原环境,影响深层砂样中的铀迁移,铀在还原环境中沉淀。
3 结论
1)研究表明,尾砂中铀的总体浸出量随时间增大而增加,直至达到最大浸出量后基本保持平衡。主要原因在于在酸浸过程中铀在尾砂中随着pH值的变化逐渐从沉淀态变为溶解态,即生成了硫酸铀酰,尽管尾矿砂经过自然风化、溶解、迁移和沉淀过程,各层尾矿砂中都含有一定量的铀。
2)通过对不同深度浅层砂中铀含量的分析,结果表明浅层尾砂中铀的浸出量与深度呈非线性关系。浅层尾矿砂中80cm处的浸出量都大于其它各层的浸出量,其主要原因在于降雨以及丰水期,铀的氧化还原等复杂的因素对铀在尾矿砂中分布的影响。此外表层尾矿砂的风化氧化作用、深层压实作用和铀本身的迁移,都会对不同层次尾矿砂中铀的分布有影响。
[1] 张 纯 .零价铁和赤铁矿去除污染水体中U(VI)的试验研究[D].湖南:南华大学,2007.
[2] Abdelouas A,Lutze W,Eric Nuttall H.Uranium contamination in the subsurface:characterization and remediation.In:Burns PC,Finch R,eds.Uranium:Mineralogy,Geochemistry and the Environment[J].Reviews in Mineralogy.Washington DC:Mineralogy Society of America.1999,33:433-473.
[3] Vandenhove H,Cuypers A Van Hees M.Oxidative stress reactions induced in beans (Phaseolus vulgaris)following exposure to uranium[J].Plant Physiol Biochem 2006,44:795-805.
[4] Gavrilescu M,Pavel LV,Cretescu I.Characterization and remediation of soils contaminated with uranium[J].J Hazard Mater,2009,163:475-510.
[5] Mason C F V,Turney W R J R,Thomson B M.Carbonate leaching of uranium from contaminated soils[J].Environ Sci Technol,1997,31:2707-2711.
[6] Rufyikiri G,Huysmans L,Wannijn J.Arbuscular mycorrhizal fungi can decrease the uptake of uranium by subterranean clover grown at high levels of uranium in soil[J].Environ Pol-lut,2004,130:427-436.
[7] Carvalho I G,Cidu R,Fanfani L.Environmental impact of uranium mining and ore processing in the Lagoa Real District,Bahia,Brazil[J].Environ Sci Technol,2005,39:8646-8652.
[8] 罗上庚 .放射性废物处理与处置[M].北京:中国环境科学出版社,2007:27-29.
[9] 吴晓燕,熊正为,彭小勇 .降雨对尾矿库地下水中核素迁移影响的模拟研究[J].安全与环境学报,2013,13(1):92-95.
[10] Gustavo J V,Cleveland J D,Arokiasamy J F.Bioreduction of U(VI)-Phthalate to a Polymeric U(IV)一 Phthalate Colloid[J].Inorg Chem,2009.48(19):9485-9490.
[11] Dmckay L,Sanford W E,Strong J M.Field—Scale Migration of Colloidal Tracers in a Fractured Shale Sap—Rolite[J].Ground Water,2000,38(1):139-147.
[12] 史维浚 .铀水文地球化学原理[M].北京:原子能出版社,1990:147-258.
[13] 曹存存,吕俊文,夏良树 .土壤胶体对渗滤液中铀(VI)迁移影响的研究进展[J].核化学与放射化学,2012,34(1):1-6.
[14] 高 柏,杜 洋,孙占学 .十红滩砂岩型铀矿床南段地浸条件分析[J].中国矿业,2013,22(6):89-92.
猜你喜欢
有色冶金设计与研究(2022年3期)2022-07-12
昆钢科技(2022年2期)2022-07-08
建材发展导向(2021年20期)2021-11-20
矿产综合利用(2021年4期)2021-10-12
有色金属(矿山部分)(2021年4期)2021-08-30
矿产综合利用(2020年1期)2020-07-24
劳动保护(2018年8期)2018-09-12
中国军转民(2017年9期)2017-12-19
新疆钢铁(2015年2期)2015-11-07
有色金属科学与工程(2015年2期)2015-05-11
