
反应pH值对微波法合成的Mo修饰的Pt/C催化剂结构和乙醇电氧化催化性能的影响
2014-06-23 06:52:08原鲜霞夏小芸马紫峰
物理化学学报 2014年5期
马 忠 原鲜霞 夏小芸 杜 娟 李 琳 马紫峰
(上海交通大学化学工程系,上海200240)
1 引言
质子交换膜燃料电池因其极高的能量密度和低温运行特性等优点近年来备受关注.1,2鉴于所用常规燃料——氢气的生产、储存和运输等问题,液体醇类(如甲醇、乙醇等)成为替代燃料之一.其中,甲醇存在一定的毒性和不稳定性,且乙醇具有更高的能量密度.3因此,直接乙醇燃料电池(DEFC)已成为低温燃料电池的研究热点.4-6
Pt催化剂已被广泛用于直接乙醇燃料电池中作为乙醇氧化的催化剂.3,7-9然而,该类催化剂尽管具备优秀的催化活性,但其表面很容易吸附乙醇氧化过程中产生的中间产物类CO物种,从而发生中毒现象而失去活性.10同时,昂贵的成本也是制约其大规模应用的主要原因之一.因此,开发低成本、高效率的乙醇电氧化催化剂已成为DEFC方向的一个重要任务.为此,多种金属如Ir、Mo、Os、Ru、Sn、W和Zr等已被引入Pt催化剂形成二元、三元甚至多元的Pt基催化剂,并对乙醇的电催化氧化表现出良好的性能.10-15其中,Mo元素尽管没有电催化乙醇的作用,但具备较好的催化一氧化碳氧化的作用,16,17Samjeské等18利用电化学质谱发现Mo元素对弱吸附态的CO具有很好的氧化作用.
微波法具有快速、高效、均匀等优点,可用于制备颗粒细小、均匀的纳米粒子,已被广泛应用于有机合成及无机材料制备等方面.19-21另据文献报道,前驱体溶液的pH值对所制得催化剂的粒径有重要影响,从而影响到催化剂的催化活性.22,23Chu等24采用微波法制备了PtRu/C催化剂,发现随着pH值的增加,催化剂颗粒的粒径逐渐变小.Zheng等25的研究表明,pH值为7时利用微波法制得的Ru85Se15/MWCNTs催化剂颗粒分散最均匀.
因此,本文采用微波法,通过调节不同反应pH值制备了一系列Mo修饰的Pt/C催化剂,对比考察了反应pH值对所制得的催化剂的微观结构、形貌及乙醇电氧化催化性能的影响,并对研究结果进行了讨论.
2 实验部分
2.1 催化剂的制备
Mo修饰的Pt/C催化剂采用微波法制备,具体合成方法为:将浓度分别为0.0488和0.0386 mol·L-1的H2PtCl6(国药,分析纯)和Na2MoO4(国药,分析纯)的乙二醇溶液加入到Vulcan XC-72(Cabot,美国)的乙二醇(国药,分析纯)浆液中,超声混合均匀后利用HCl(国药,分析纯)和NaOH(国药,分析纯)调节混合液的pH值(本文选取1、8、12和14四个pH值作为考察对象),然后将之置于微波反应器(Sanle WP650D)中,在功率为325 W、冷凝回流的状态下反应15 min,然后洗去氯离子,真空干燥后得产物.
本文所制备的Mo修饰的Pt/C催化剂中,Pt:Mo的摩尔比均为2:1,Pt的质量分数为20%(w).
2.2 催化剂的表征
催化剂的微观形貌采用日本电子株式会社的JEOL JEM 2000 EX透射电子显微镜(TEM)进行观察.
催化剂的晶体结构采用日本理学Rigaku D/MAX PC 2000型X射线衍射(XRD)仪器进行表征,测试条件为:CuKa射线源,管电压为35 kV,管电流为25 mA,扫描速率为6(°)·min-1.
催化剂的表面形态采用美国PHI公司的PHI 5000C ESCA System仪器进行测试.测试条件为铝/镁靶,高压14.0 kV,功率250 W,通能93.9 eV,真空优于1.33×10-6Pa.采用美国RBD公司的RBD147数据采集卡和AugerScan 3.21软件分别采集样品的0-1200(1100)eV的全扫描谱,而后采集各元素相关轨道的窄扫描谱.以C 1s(284.5 eV)为基准进行结合能校正.
2.3 催化剂的电化学性能表征
催化剂对乙醇电氧化过程的催化性能用循环伏安曲线、计时电流法和电化学阻抗谱进行测试.测试采用常规的三电极电解池,其中以覆有所制备催化剂的玻碳电极为工作电极、铂丝为对电极、Hg/Hg2SO4电极为参比电极、0.5 mol·L-1H2SO4和 0.5 mol·L-1H2SO4+0.5 mol·L-1C2H5OH为电解液.工作电极的具体制备方法为:取5 mg催化剂,加入一定配比的水和5%(w)的Nafion乳液配成5 g·L-1的催化剂悬乳液,超声分散10 min后移取10 μL涂于有效面积为0.096 cm2的玻碳电极表面,干燥后得到工作电极.循环伏安测试的电位扫描速率为50 mV·s-1;计时电流测试采用恒电位0.273 V(vsHg/Hg2SO4),时间为1000 s;交流阻抗测试采用美国阿美特克公司的Solartron SI1287电化学工作站进行,频率扫描范围为100 kHz-1 Hz,交流电压振幅为5 mV,测试电极电位为0.8 V.
3 结果与讨论
图1为不同反应pH值所制备的Mo修饰Pt/C催化剂的TEM图.图2为TEM图中催化剂组分颗粒对应的粒径分布图.从图1和图2可以看出,当pH值为1时,负载在碳载体上的催化剂颗粒数目较少、粒径较大且分布范围较广,说明强酸环境不利于活性组分在碳载体上的均匀负载;当提高pH值为8时,催化剂颗粒的数目有所增加,但颗粒尺寸仍旧较大且粒径分布较广;当pH值进一步提升为12时,碳载体上催化剂组分的颗粒较细小,但局部有所团聚,不利于催化剂活性的充分发挥;当提高pH值为14时,催化剂组分在碳载体上分布均匀、且粒径细小.这些结果表明,反应pH值对微波法合成的催化剂中催化活性组分的粒径大小与分布具有可控性的调节作用,强碱环境有利于粒径细小的催化活性组分在碳载体上的均匀分布.这可能与催化剂合成过程中乙二醇氧化的产物乙醇酸和乙醇酸盐有关.其中,乙醇酸盐对金属离子具备很强的稳定作用,能够减缓金属在碳载体上的沉积,从而使得形成的金属颗粒尺寸细小并均匀分散.23当溶液的pH值较高时,溶液中乙醇酸盐的浓度也较高,因而有利于制得粒径细小且均匀分布的金属粒子.当溶液的pH值较低时,溶液中乙醇酸盐的浓度较低,乙醇酸的浓度较高.而乙醇酸对金属粒子的稳定作用很微弱,因而制备的金属粒子的粒径较大、分布也不均匀.

图1 不同反应pH值制得的Mo修饰的Pt/C催化剂的TEM图Fig.1 TEM images of Mo-modified Pt/C catalysts synthesized at various pH values
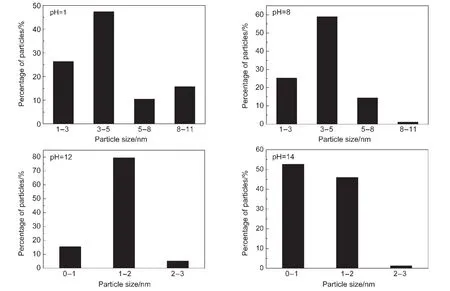
图2 不同反应pH值制得的Mo修饰的Pt/C催化剂的粒径分布图Fig.2 Particle size distributions in the Mo-modified Pt/C catalysts synthesized at various pH values
图3为不同反应pH值制得的Mo修饰的Pt/C催化剂的XRD谱图.可以看出,所有催化剂的XRD谱图中在24.9°附近均有碳C(002)面的衍射峰,且都有明显的Pt的衍射特征峰,但都没有Mo的特征峰.这可能是由于催化剂中Mo元素以无定形型态存在或已嵌入到Pt的晶格中.26当pH值为1时,所制得的Mo修饰的Pt/C催化剂的Pt(111)的特征峰比较明显,其他峰强度则较弱或不明显.说明当反应溶液呈酸性时,所制备的催化剂中Pt的结晶度较差.当pH值为8、12或14时,所制得的催化剂中面心立方晶格结构的Pt的四个特征衍射峰Pt(111)、(200)、(220)和(311)均很明显,说明碱性环境有利于制备结晶度完好的Pt颗粒.同时,通过比较可以发现,Mo修饰的Pt/C催化剂中Pt的晶面有所负移,说明掺杂的Mo嵌入到了Pt的晶格当中并使其晶格参数增大.通常,Pt(111)、(200)和(311)晶面的衍射峰强度会受到载体碳材料特征峰的影响,而Pt(220)晶面特征峰不受载体干扰.27从图中可以看出,随着pH值的增加,Pt(220)晶面衍射峰的半峰宽依次增大,说明催化剂中活性组分的晶粒逐渐变小,这与TEM观察到的结果是一致的.

图3 不同反应pH值制得的Mo修饰的Pt/C催化剂的XRD谱图Fig.3 XRD patterns of Mo-modified Pt/C catalysts synthesized at various pH values pH:(a)1;(b)8;(c)12;(d)14
图4为不同反应pH值制得的Mo修饰的Pt/C催化剂中Pt元素4f轨道的XPS谱图,其中71.0-71.5 eV(4f7/2轨道)和74.3-74.8 eV(4f5/2轨道)的峰对应单质金属Pt,72.4-72.7 eV(4f7/2轨道)和75.5-75.9 eV(4f5/2轨道)对应Pt(II),可能为PtO或Pt(OH)2.对XPS谱图的拟合结果(见图4)表明,各催化剂中的Pt主要以单质金属的形式存在,二价Pt只占很小的一部分,由此获得的不同价态Pt的结合能列于表1.可以看出,随着pH值的增加,无论是4f7/2还是4f5/2轨道,Pt(0)的结合能位置都逐渐正移,这可能是由于随着反应pH值的增加,所合成的催化剂中Mo元素和Pt元素之间的作用力逐渐增强,28也进一步证明了在所合成的催化剂中Mo已经嵌入到Pt的晶格当中.
图5为不同反应pH值制得的Mo修饰的Pt/C催化剂在0.5 mol·L-1H2SO4溶液中的循环伏安曲线,由此计算得到的各催化剂中Pt的有效电化学比表面积(ECSA)列于表2.可以看出,随着反应pH值的增大,催化剂的有效电化学比表面积明显增加,pH值为14时达到最大值(41.91 m2·g-1).结合前文对TEM结果的讨论,这可能是因为催化剂的颗粒随pH值增加而减小导致的.
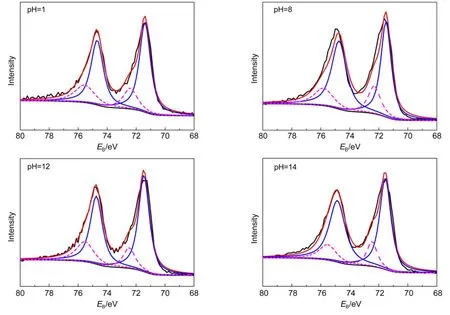
图4 不同反应pH值制得的Mo修饰的Pt/C催化剂中Pt的XPS谱图Fig.4 XPS spectra of Pt in Mo-modified Pt/C catalysts synthesized at various pH values
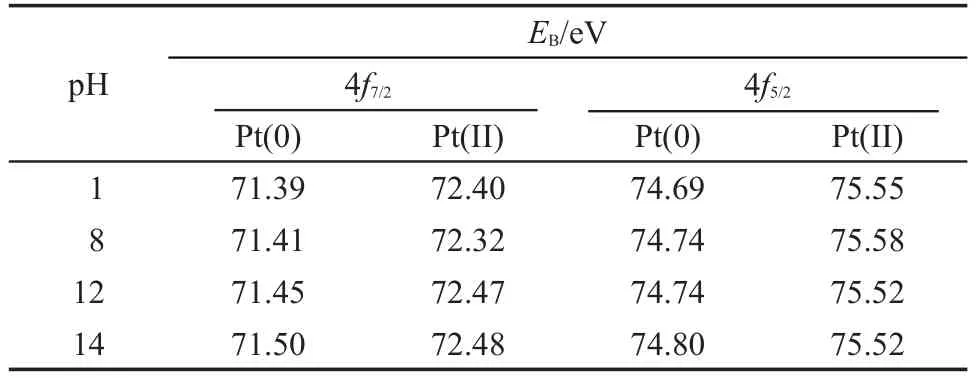
表1 不同反应pH值制得的Mo修饰的Pt/C催化剂中不同价态Pt的结合能Table 1 Binding energy of Pt species in Mo-modified Pt/C catalysts synthesized at various pH values
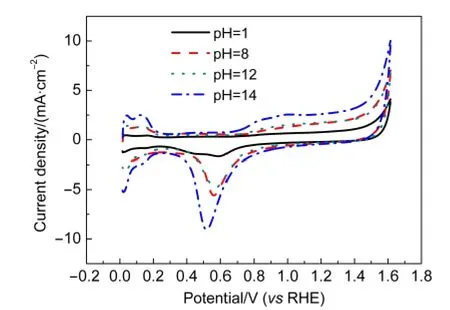
图5 不同反应pH值制得的Mo修饰的Pt/C催化剂在0.5 mol·L-1硫酸溶液中的循环伏安曲线Fig.5 CV curves of Mo-modified Pt/C catalysts synthesized at various pH values in 0.5 mol·L-1 H2SO4solution
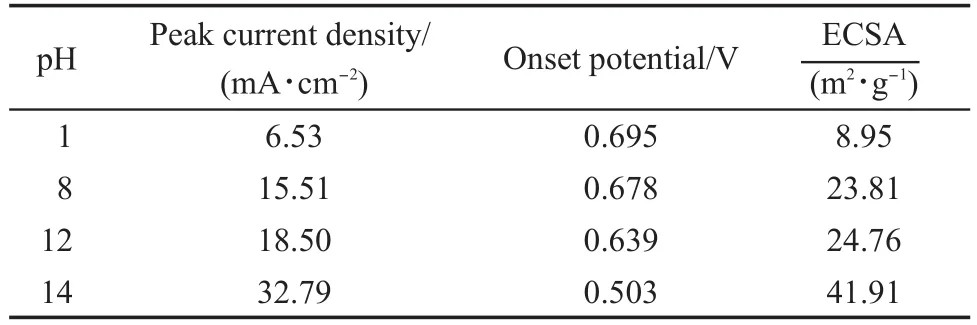
表2 不同反应pH值制得的Mo修饰的Pt/C催化剂的乙醇电氧化催化性能Table 2 Electro-catalytic performance of Mo-modified Pt/C catalysts synthesized at various pH values towards ethanol oxidation

图6 不同反应pH值制备的Mo修饰的Pt/C在0.5 mol·L-1硫酸+0.5 mol·L-1乙醇溶液中的循环伏安曲线Fig.6 CV curves of Mo-modified Pt/C catalysts synthesized at various pH values in 0.5 mol·L-1H2SO4+0.5 mol·L-1C2H5OH solution
图6为不同反应pH值制得Mo修饰的Pt/C催化剂在0.5 mol·L-1H2SO4+0.5 mol·L-1C2H5OH溶液中的循环伏安曲线,其中各催化剂对乙醇电催化氧化反应的起始电位(onset电位,取反应电流达到2 mA·cm-2时的电位)和峰值电流密度列于表2.可以看出,随着反应pH的提高,所得催化剂对乙醇电催化氧化反应的起始电位逐渐降低、峰值电流密度逐渐增大,当pH值提高到14时,其乙醇氧化起始电位和峰值电流密度分别达到为0.503 V和32.79 mA·cm-2,这说明溶液pH值的提高有利于制备乙醇氧化催化活性更高的Mo修饰的Pt/C催化剂.
不同反应pH值制得的Mo修饰的Pt/C催化剂在0.5 mol·L-1H2SO4+0.5 mol·L-1C2H5OH溶液中的计时电流曲线如图7所示.在本文所研究的时间范围内,各催化剂所表现出来的乙醇电催化氧化电流随反应pH值的增加而增大.在1000 s时,反应pH值为1、8、12、14所制得的催化剂的电流密度分别为0.94、3.16、2.06和7.07 mA·cm-2.这些结果说明反应pH值为14时制得的催化剂对乙醇的电催化氧化不仅具有最高的催化活性,而且具有最好的稳定性.

图7 不同反应pH值制得的Mo修饰的Pt/C催化剂在0.5 mol·L-1硫酸+0.5 mol·L-1乙醇溶液中的计时电流曲线Fig.7 Chronoamperometry curves of Mo-modified Pt/C catalysts synthesized at various pH values in 0.5 mol·L-1H2SO4+0.5 mol·L-1C2H5OH solution

图8 不同反应pH值制得的Mo修饰的Pt/C催化剂在0.5 mol·L-1硫酸+0.5 mol·L-1乙醇溶液中的电化学阻抗谱Fig.8 Electrochemical impedance spectroscopy of Mo-modified Pt/C catalysts synthesized at various pH values in 0.5 mol·L-1H2SO4+0.5 mol·L-1C2H5OH solution
图8为不同反应pH值制得的Mo修饰的Pt/C催化剂的电化学阻抗谱图.可以看出,乙醇在反应pH值为14时所制备的Mo修饰的Pt/C催化剂上的电化学反应电阻最小,说明在该催化剂上乙醇的电氧化反应具备最快的动力学过程.当pH值依次降低到12、8和1时,在所制备的催化剂上,乙醇的电化学反应电阻依次增大,且pH值为1时制得的Mo修饰的Pt/C催化剂上的电化学反应电阻远大于其它条件下制备的催化剂,说明乙醇在Mo修饰的Pt/C催化剂上的电催化氧化反应随催化剂制备过程中反应pH值的增加而变得更容易,催化剂的活性更高.
综上所述,当采用微波法制备Mo修饰的Pt/C催化剂时,增加反应的pH值有利于提高所制得催化剂的乙醇电氧化催化活性和稳定性.这不仅与催化剂中Pt元素和Mo元素之间存在的协同作用相关,28,29而且催化剂中活性组分Pt颗粒的尺寸以及分散状态也是可能的原因之一,细小Pt颗粒在载体表面的均匀分布有利于催化剂性能的改善.
4 结论
反应pH值对微波法合成的Mo修饰的Pt/C催化剂作为乙醇电氧化反应催化剂的微结构、形貌和催化性能有重要的影响.反应pH值的提高有利于形成结晶良好的、小颗粒均匀分散的催化剂,从而改善催化剂的乙醇电氧化催化性能.在本文的研究范围内,当反应pH值为14时,所制备的Mo修饰的Pt/C催化剂颗粒细小,且均匀分布在碳载体的表面,表现出最好的乙醇电氧化催化活性和稳定性.
(1) Wee,J.H.Renew.Sust.Energ.Rev.2007,11,1720.doi:10.1016/j.rser.2006.01.005
(2) Peighambardoust,S.J.;Rowshanzamir,S.;Amjadi,M.Int.J.Hydrog.Energy2010,35,9349.doi:10.1016/j.ijhydene.2010.05.017
(3) Zhou,W.;Zhou,Z.;Song,S.;Li,W.;Sun,G.;Tsiakaras,P.;Xin,Q.Appl.Catal.B-Environ.2003,46,273.doi:10.1016/S0926-3373(03)00218-2
(4) Garcia,A.;Linares,J.;Chatenet,M.;Ticianelli,E.Electrocatalysis2014,5,41.doi:10.1007/s12678-013-0162-1
(5) Kamarudin,M.Z.F.;Kamarudin,S.K.;Masdar,M.S.;Daud,W.R.W.Int.J.Hydrog.Energy2013,38,9438.doi:10.1016/j.ijhydene.2012.07.059
(6) Song,S.;Tsiakaras,P.Appl.Catal.B-Environ2006,63,187.doi:10.1016/j.apcatb.2005.09.018
(7) Rightmire,R.A.;Rowland,R.L.;Boos,D.L.;Beals,D.L.J.Electrochem.Soc.1964,111,242.doi:10.1149/1.2426092
(8) Lai,S.C.S.;Koper,M.T.M.Phys.Chem.Chem.Phys.2009,11,10446.doi:10.1039/b913170a
(9) Gomes,J.F.;Bergamaski,K.;Pinto,M.F.S.;Miranda,P.B.J.Catal.2013,302,67.doi:10.1016/j.jcat.2013.02.024
(10) Antolini,E.J.Power Sources2007,170,1.doi:10.1016/j.jpowsour.2007.04.009
(11)Almeida,T.S.;Palma,L.M.;Morais,C.;Kokoh,K.B.;De Andrade,A.R.J.Electrochem.Soc.2013,160,F965.
(12) Linares,J.;Zignani,S.;Rocha,T.;Gonzalez,E.J.Appl.Electrochem.2013,43,147.doi:10.1007/s10800-012-0496-z
(13)Antolini,E.ChemSusChem2013,6,966.doi:10.1002/cssc.v6.6
(14) Hou,Z.;Yi,B.;Yu,H.;Lin,Z.;Zhang,H.J.Power Sources2003,123,116.doi:10.1016/S0378-7753(03)00515-9
(15) Tayal,J.;Rawat,B.;Basu,S.Int.J.Hydrog.Energy2011,36,14884.doi:10.1016/j.ijhydene.2011.03.035
(16) Santiago,E.I.;Camara,G.A.;Ticianelli,E.A.Electrochim.Acta2003,48,3527.doi:10.1016/S0013-4686(03)00474-2
(17) Ioroi,T.;Fujiwara,N.;Siroma,Z.;Yasuda,K.;Miyazaki,Y.Electrochem.Commun.2002,4,442.doi:10.1016/S1388-2481(02)00341-7
(18) Samjeské,G.;Wang,H.;Löffler,T.;Baltruschat,H.Electrochim.Acta2002,47,3681.doi:10.1016/S0013-4686(02)00338-9
(19) Zeng,X.;Yuan,X.X.;Xia,X.Y.;Du,J.;Zhang,H.J.;Ma,Z.F.J.Inorg.Mater.2010,25,359.[曾 鑫,原鲜霞,夏晓芸,杜 娟,张慧娟,马紫峰.无机材料学报,2010,25,359.]doi:10.3724/SP.J.1077.2010.00359
(20) Du,J.;Yuan,X.X.;Yu,J.H.;Ma,Z.F.Chin.J.Power Sources2007,31,873.[杜 娟,原鲜霞,余江虹,马紫峰.电源技术,2007,31,873.]
(21)Wu,X.;Jiang,Q.Z.;Ma,Z.F.;Shangguan,W.F.Chin.J.Inorg.Chem.2006,22,341.[吴 省,蒋淇忠,马紫峰,上官文峰.无机化学学报,2006,22,341.]
(22)Wang,X.Z.;Zheng,J.S.;Fu,R.;Ma,J.X.Acta Phys.-Chim.Sin.2011,27,85.[王喜照,郑俊生,符 蓉,马建新.物理化学学报,2011,27,85.]doi:10.3866/PKU.WHXB20110111
(23)Bock,C.;Paquet,C.;Couillard,M.;Botton,G.A.;MacDougall,B.R.J.Am.Chem.Soc.2004,126,8028.doi:10.1021/ja0495819
(24)Chu,Y.Y.;Wang,Z.B.;Jiang,Z.Z.;Gu,D.M.;Yin,G.P.Fuel Cells2010,10,914.doi:10.1002/fuce.v10.6
(25) Zheng,Q.;Cheng,X.;Jao,T.C.;Weng,F.B.;Su,A.;Chiang,Y.C.Int.J.Hydrog.Energy2011,36,14599.doi:10.1016/j.ijhydene.2011.08.038
(26) Li,L.;Xu,B.Q.Acta Phys.-Chim.Sin.2005,21,1132.[李 莉,徐柏庆.物理化学学报,2005,21,1132.]doi:10.3866/PKU.WHXB20051014
(27)Zhou,W.J.;Li,W,Z.;Zhou,Z.H.;Song,S.Q.;Wei,Z.B.;Sun,G.Q.;Tsiakaras,P.;Xin,Q.Chem.J.Chin.Univ.2003,24,858.[周卫江,李文震,周振华,宋树芹,魏昭彬,孙公权,Tsiakaras,P.,辛 勤.高等学校化学学报,2003,24,858.]
(28) Yan,Z.;Wang,H.;Zhang,M.;Jiang,Z.;Jiang,T.;Xie,J.Electrochim.Acta2013,95,218.doi:10.1016/j.electacta.2013.02.031
(29)Cui,G.;Shen,P.K.;Meng,H.;Zhao,J.;Wu,G.J.Power Sources2011,196,6125.doi:10.1016/j.jpowsour.2011.03.042
猜你喜欢
物理化学学报(2024年9期)2024-09-27 00:00:00
保健与生活(2023年6期)2023-03-17 08:39:54
中国有色金属学报(2018年2期)2018-03-26 07:58:37
红领巾·探索(2018年12期)2018-01-26 12:34:14
金色年华(2017年12期)2017-07-18 11:11:20
中南大学学报(自然科学版)(2016年2期)2017-01-19 07:37:25
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年4期)2014-02-28 17:31:23
无机化学学报(2014年3期)2014-02-28 17:30:48
