
苯部分加氢制环己烯新型Ru-B/MOF催化剂
2014-06-23 06:52:04谭晓荷周功兵窦镕飞范康年乔明华宗保宁
物理化学学报 2014年5期
谭晓荷 周功兵 窦镕飞 裴 燕 范康年乔明华,* 孙 斌 宗保宁,*
(1上海市分子催化和功能材料重点实验室,复旦大学化学系,上海200433;2催化材料与反应工程国家重点实验室,中国石化石油化工科学研究院,北京100083)
1 引言
近年来,新型多孔复合材料——金属-有机骨架(MOF)材料受到了人们的广泛关注.MOF材料通过金属离子与有机配体自组装形成,具有拓扑结构多样、比表面积大、孔隙率高、孔道规则、孔道尺寸可调等优点,在气体储存与分离、1,2分子筛分与识别3及催化4-6等领域有着广阔的应用前景.在催化应用中,MOF材料通常有不饱和配位的金属中心或功能化的有机配体,从而具有一定的Lewis酸性,使MOF材料本身显示出催化作用.7,8MOF材料的有序孔道也可以在特定反应中起到择形催化的作用.9另一方面,MOF材料的大比表面积和多孔性使其有可能成为优秀的加氢催化剂载体.如Jiang等10采用沉积-还原法制备了ZIF-8(I22)负载的Au@Ag核壳催化剂,发现在NaBH4还原4-硝基苯酚反应中,催化活性高于Au和Ag的单金属催化剂.Proch等11制备了Pt@MOF-177催化剂,在无溶剂、无碱、室温下的醇氧化反应中显示出较高的活性.Schröder等12制备了Ru@MOF-5催化剂用于苯加氢制环己烷,在0.3 MPa H2压力和75°C下反应20 h,苯的转化率为25%.Wu等13在超临界CO2-甲醇流体中制备了Ru@Zr-MOF催化剂,在60°C和6 MPa反应条件下,苯加氢生成环己烷的转换频率(TOF)为5260 h-1,高于Ru/La-MOF催化剂.其原因可能是Zr-MOF同时具有微孔和介孔,有利于反应物和产物的扩散及反应的发生.
与苯加氢制环己烷相比,苯部分加氢制环己烯在热力学和动力学上难度均更高.14由于环己烯拥有活泼的C=C双键,是一种用途更广的化学合成中间体,15因此苯部分加氢制环己烯催化剂有着重要的研究价值.已有的研究表明,在苯部分加氢反应中,催化剂载体的性质对环己烯选择性的影响很大.14,16-18然而,在文献中尚未见到将MOF材料用于苯部分加氢反应的报道.通常,为了促进中间产物环己烯从催化剂表面脱附,以提高环己烯的选择性,苯部分加氢反应一般在140°C以上有水相存在的条件下进行.19因此在选择MOF材料作为苯部分加氢催化剂载体时,热稳定性尤其是水热稳定性是重要考虑因素.在保证MOF材料有较高热稳定性的前提下,优先选择以水热(溶剂热)法制备的MOF材料为催化剂载体,以期MOF材料的结构在反应中能够保持稳定.Férey与其合作者20-23最早开展了MIL(materials of Insititut Lavoisier)系列MOF的研究工作.他们通过水热法合成了大量三价金属与对苯二甲酸或均苯三甲酸配位形成的MIL-n材料.MIL-n材料通过MO4(OH)2(M=Cr3+,Al3+,Fe3+)八面体与有机配体相互桥联,形成具有菱形孔道的三维骨架结构,其中MIL-53(Al)有较高的热稳定性,其热分解温度高达500°C.22Cavka等24报道了通过溶剂热法合成的另一种高热稳定性的MOF材料,命名为UIO-66.它通过高度对称的八面体无机金属单元Zr6O4(OH)4与有机配体相互桥联,形成四面体和八面体两种类型的孔笼.每个八面体孔笼的八个面上,均与一个四面体孔笼相连,在三维空间形成不断延伸的骨架结构材料,其热分解温度亦高于500°C.
本文在上述文献工作的基础上,制备了多种以MOF材料为载体的负载型非晶态Ru-B合金催化剂,首次考察了它们在苯部分加氢反应中的催化性能.对催化性能比较典型的两个催化剂的物理化学性质进行了表征,初步探讨了苯部分加氢性能与Ru-B/MOF催化剂结构之间的关系,并对反应温度和压力的影响进行了考察.
2 实验部分
2.1 实验药品
九水合硝酸铝(Al(NO3)3·9H2O),六水合氯化铝(AlCl3·6H2O),九水合硝酸铬(Cr(NO3)3·9H2O),硼氢化钾(KBH4),乙醇胺(HOCH2CH2NH2,ETA),对苯二甲酸(C8H6O3,H2BDC),七水合硫酸锌(ZnSO4·7H2O),异丙醇胺(H2NCH2CH(OH)CH3,IPTA),甘油(HOCH2CH(OH)CH2OH,GL),乙二胺(H2NCH2CH2NH2,EDA),国药集团化学试剂有限公司;均苯三甲酸(C9H6O6,H3BTC),氯化锆(ZrCl4),上海晶纯生化科技股份有限公司;浓硝酸(HNO3),太仓市直塘化工有限公司;乙醇,上海振兴化工一厂;N,N-二甲基甲酰胺(DMF),江苏强盛功能化学股份有限公司;氢氟酸(HF,40%),江苏彤晟化学试剂有限公司;苯(C6H6),宝钢集团有限公司;三水合三氯化钌(RuCl3·3H2O),上海嶅稞实业有限公司;2-氨基对苯二甲酸(C8H7NO4,H2ATA),铁粉(Fe),1,3-丙二胺(H2NCH2CH2CH2NH2,1,3-PDA),美国Sigma-Aldrich公司;以上试剂均为分析纯.
2.2 催化剂制备
MIL-53(Al):22将20.80 g Al(NO3)3·9H2O、4.61 g H2BDC、80 mL水依次加入有聚四氟乙烯内衬的水热釜中,搅拌下超声30 min,使Al(NO3)3·9H2O充分溶解,并使H2BDC在溶液中均匀分散.密封,在220°C下水热3天.然后冷却至室温,抽滤,用去离子水洗涤,60°C下干燥过夜,得到白色粉末,记为MIL-53(Al)-as.采用DMF溶剂热法25除去MIL-53(Al)-as孔中未反应的H2BDC.步骤如下:将1.00 g MIL-53(Al)-as分散在25 mL DMF中,于150°C下溶剂热处理15 h.冷却,抽滤,60°C下干燥过夜,在马弗炉中280°C焙烧12 h,升温速率5°C·min-1,得到MIL-53(Al).
MIL-53(Cr):21将10.00 g Cr(NO3)3·9H2O、4.15 g H2BDC、1.25 g HF和126 mL水依次加入有聚四氟乙烯内衬的水热釜中,搅拌下超声30 min,使Cr(NO3)3·9H2O充分溶解,并使H2BDC在溶液中均匀分散.密封,在220°C下水热3天.冷却至室温,抽滤,用去离子水洗涤,60°C下干燥过夜.将所得浅绿色粉末采用DMF溶剂热法25除去孔中未反应的H2BDC,具体步骤同MIL-53(Al).冷却,抽滤,60°C下干燥过夜,在马弗炉中200°C焙烧12 h,升温速率5°C·min-1,得到MIL-53(Cr).
MIL-101(Cr):26将5.20 g Cr(NO3)3·9H2O、2.16 g H2BDC、0.74 g HF和62 mL水依次加入有聚四氟乙烯内衬的不锈钢水热釜中,溶解后在220°C水热8 h.冷却至室温,抽滤,并用去离子水洗涤.将所得固体采用DMF溶剂热法25除去孔中未反应的H2BDC,具体步骤同MIL-53(Al).冷却,抽滤,洗涤,所得浅绿色固体在150°C干燥12 h,得到MIL-101(Cr).
MIL-100(Fe):27将 1.00 g铁粉、2.50 g H3BTC、1.75 g HF、1.00 g HNO3和89 mL水依次加入有聚四氟乙烯内衬的不锈钢水热釜中,溶解后在150°C下水热12 h.冷却,抽滤,并用去离子水洗涤.所得固体经过下列步骤纯化:先分散在400 mL水中,80°C回流5 h,抽滤.再分散到200 mL乙醇中,60°C回流3 h,抽滤,洗涤至滤液无色.所得浅橘黄色固体在60°C下干燥过夜,得到MIL-100(Fe).
UIO-66(Zr):28将0.16 g ZrCl4、0.10 g H2BDC、12 μL H2O和78 mL DMF依次加入有聚四氟乙烯内衬的不锈钢水热釜中,溶解后在120°C溶剂热24 h.冷却,抽滤,将所得固体分散在20 mL DMF中,振荡4 h后离心,再分散到20 mL乙醇中,振荡4 h后离心.所得白色固体在60°C干燥过夜,得到UIO-66(Zr).
MIL-53(Al)-NH2:29将4.94 g AlCl3·6H2O、3.76 g H2ATA和50 mL水依次加入有聚四氟乙烯内衬的水热釜中,搅拌下超声30 min,使AlCl3·6H2O充分溶解,并使H2ATA在溶液中均匀分散.密封,在150°C下水热5 h.然后冷却至室温,抽滤,用去离子水洗涤,60°C下干燥过夜.得到的黄色粉末采用DMF溶剂热法25除去孔中未反应的H2ATA,具体步骤同MIL-53(Al).冷却,抽滤,60°C下干燥过夜,在马弗炉中130°C焙烧12 h,升温速率5°C·min-1,得到MIL-53(Al)-NH2.
UIO-66(Zr)-NH2:30将0.45 g ZrCl4、0.35 g H2ATA、34.7 μL H2O和73 mL DMF依次加入有聚四氟乙烯内衬的不锈钢水热釜中,溶解后在120°C下溶剂热24 h.其他步骤与UIO-66(Zr)相同,但所用DMF和乙醇均为56 mL.
采用浸渍-化学还原法制备负载型非晶态Ru-B合金催化剂.将0.5 mL 0.4 mol·L-1的RuCl3·3H2O水溶液滴加到0.17 g上述MOF材料中,再加入0.4 mL水.超声30 min后,向上述悬浊液中滴加0.8 mL 1.0 mol·L-1的KBH4水溶液进行还原(KBH4/Ru摩尔比为4:1),还原在冰水浴中进行.待反应至无气泡产生后,用去离子水洗涤催化剂,至滤液中无氯离子(AgNO3溶液检测),即得非晶态Ru-B/MOF催化剂.
2.3 催化剂表征
催化剂体相组成采用电感耦合等离子体原子发射光谱(ICP-AES,美国Thermo Elemental公司IRISIntrepid)测定.Brunauer-Emmett-Teller比表面积(SBET)及孔性质采用N2物理吸附在美国Quantachrome公司AUTOSORB-1MP型物理吸附仪上测定.吸附前,样品于N2(99.9%,上海浦江特种气体有限公司)气氛下300°C预处理10 h.
催化剂的活性比表面积(SRu)采用CO脉冲吸附在美国Micromeritics 2750型化学吸附仪上测定.将约40 mg样品装入样品管,在高纯He(99.999%,上海尤嘉利液氦有限公司)气氛中于200°C预处理2 h,然后降温至30°C.在该温度下CO(99.9%,上海尤嘉利液氦有限公司)脉冲进样,31至热导检测器(TCD)检出的峰面积恒定为止,即达到吸附饱和.根据吸附面积计算SRu时,假定吸附的CO与Ru原子的化学计量比为0.6:1,31且Ru的表面原子密度为1.63×1019atoms·m-2.32
粉末X射线衍射(XRD)在德国BrukerAXS公司D8 Advance型衍射仪上测定.CuKα线为激发源(λ=0.15418 nm),石墨单色器,管电压40 kV,管电流40 mA,扫描速率4(°)·min-1.傅里叶变换红外(FTIR)光谱在美国Nicolet公司Nexus 470型红外光谱仪上采集,KBr压片制样,扫描范围4000-400 cm-1,仪器分辨率4 cm-1,扫描32次.热重分析(TGA)在美国Perkin-Elmer公司Series型热重分析仪上进行.在空气气氛下加热,加热速率5°C·min-1,气体流速20 mL·min-1.
X射线光电子能谱(XPS)在美国Perkin-Elmer公司PHI5000C型能谱仪上采集.MgKα线(hν=1253.6 eV)为激发源,电压14.0 kV,功率250 W,通能93.90 eV,X射线与样品夹角54°.将乙醇保护的催化剂转移至预处理室,室温下真空脱气过夜,然后转移至分析室,背景压力优于2×10-7Pa.
催化剂的形貌、Ru-B的粒径及选区电子衍射(SAED)采用日本JEOL生产的JEM2011型透射电子显微镜(TEM)观察,工作电压200 kV.将催化剂分散于无水乙醇中,超声10 min,然后滴到表面覆有碳膜的铜网上.在TEM照片中量取约200个粒子的粒径,绘制粒径分布图,进行Gauss函数拟合,计算平均粒径和标准偏差.
RuK边X射线吸收谱(XAS)采用透射模式在北京高能所同步辐射实验室(BSRF)的1W1B线站上测定.吸收边能量22117 eV,电子束能量2.5 GeV,电流200 mA.将乙醇保护的催化剂压到铝窗中,置于样品台上.采用NSRLXAFS 3.0软件包对数据进行处理,33以获得X射线吸收近边结构(XANES)和扩展X射线吸收精细结构(EXAFS)数据.为分析非晶态Ru-B合金的无序结构,采用非对称配位分布函数为Gaussian函数和指数函数的卷积的修正EXAFS公式对结构参数进行模拟计算.34分别以六方密堆积(hcp)Ru和Ru2B3为模型化合物,采用FEFF8.20软件计算Ru-Ru和Ru-B的振幅函数和相移函数.35
2.4 催化性能评价
液相苯部分加氢反应在带有石英衬套的机械搅拌不锈钢高压反应釜(CJF-05,大连通达反应釜厂)中进行,容积500 mL.在釜中加入0.19 g催化剂,100 mL水,50 mL苯及0.5 mL乙醇胺.密闭后,用H2(99.5%,上海比欧西气体有限公司)置换5次以除尽釜内空气.于3.0 MPa H2压力下加热,升温至反应温度(140-190°C)后,调节至所需H2压力(4-6 MPa),开启搅拌至转速为1000 r·min-1,以消除扩散效应,16同时开始计时.反应过程中定时取样,在GC122型气相色谱仪(上海分析仪器厂)上分析产物组成.色谱分析条件为:PEG-20M填充柱,TCD检测,H2载气,色谱柱、检测器及进样器温度均为130°C,桥电流100 mA.
苯转化率(X)、环己烯选择性(SCHE)及环己烯得率(YCHE)分别根据以下公式计算:

上式中,下标0和t分别对应反应时间为0及反应时间为t时苯或环己烯的浓度.
催化剂的活性用初始反应速率(r0)表示.r0通过拟合苯的浓度-时间曲线,微分后外推至反应时间为0时得到,表示反应初始单位时间单位质量催化剂上转化的苯的量(mmol·min-1·g-1).环己烯初始选择性(S0)通过外推环己烯选择性-时间曲线至反应时间为0时得到.
3 结果与讨论
3.1 苯部分加氢反应性能
在典型的苯部分加氢反应条件下,考察了7种Ru-B/MOF催化剂的催化性能.需要指出的是,为提高环己烯的选择性,在苯部分加氢的反应液中需要引进修饰剂.36我们以Ru-B/MIL-53(Al)催化剂为代表,对无机、有机修饰剂进行了筛选,结果见图1.虽然ZnSO4对氧化物负载的Ru催化剂的选择性的促进作用最为突出,19但我们的实验表明其对Ru-B/MOF催化剂的修饰效果不佳.我们发现乙醇胺对Ru-B/MOF催化剂的修饰效果最好,环己烯选择性S0和得率均最高,故本文采用乙醇胺作为修饰剂.
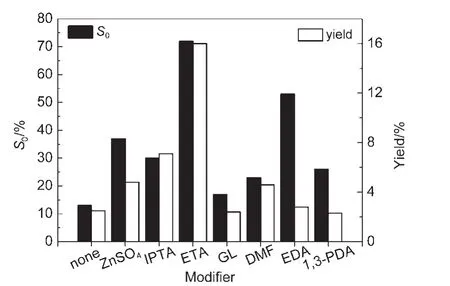
图1 修饰剂对Ru-B/MOF催化剂苯部分加氢性能的影响Fig.1 Effect of various modifiers on the partial hydrogenation of benzene over the Ru-B/MOF catalyst
表1给出了Ru-B/MOF催化剂上的反应结果.由表1可见,催化活性r0的顺序为Ru-B/MIL-53(Al)>Ru-B/MIL-53(Al)-NH2>Ru-B/UIO-66(Zr)>Ru-B/UIO-66(Zr)-NH2>Ru-B/MIL-53(Cr)>Ru-B/MIL-101(Cr)>>Ru-B/MIL-100(Fe),选择性S0的顺序为Ru-B/MIL-53(Al)≈Ru-B/MIL-53(Cr)>Ru-B/UIO-66(Zr)-NH2>Ru-B/MIL-101(Cr)>Ru-B/MIL-53(Al)-NH2>Ru-B/UIO-66(Zr)≈Ru-B/MIL-100(Fe).其中活性最高的 Ru-B/MIL-53(Al)催化剂的r0为 23 mmol·min-1·g-1,与Zhou等32报道的Ru-B/ZrO2催化剂的活性接近.Ru-B/MIL-100(Fe)催化剂的r0最低,仅为0.4 mmol·min-1·g-1.同时,环己烯选择性最高的Ru-B/MIL-53(Al)和Ru-B/MIL-53(Cr)催化剂的S0可达72%,高于Schwab等37报道的以Al2O3为载体的Ru催化剂上的环己烯初始选择性(50%-60%).Ru-B/UIO-66(Zr)和Ru-B/MIL-100(Fe)催化剂的S0最低,为45%.综合考虑活性和选择性,在所考察的MOF材料中,MIL-53(Al)最适合作为苯部分加氢催化剂的载体,而MIL-100(Fe)效果最差.
表1也表明,不仅MOF的种类(MIL或UIO)能够影响苯部分加氢催化剂的性能,而且对于相同结构的MOF材料,配位离子也会对催化性能产生极大的影响(Entries 1-4).此外,对于不同的MOF材料,NH2基修饰对催化性能的影响也有所不同.与Ru-B/MIL-53(Al)催化剂相比,Ru-B/MIL-53(Al)-NH2催化剂的活性变化不大,但选择性降低较明显.与Ru-B/UIO-66(Zr)相比,Ru-B/UIO-66(Zr)-NH2催化剂活性降低,但选择性明显改善.这些初步研究结果证明了MOF材料用作苯部分加氢催化剂的载体可行性.通过深入系统的研究,有望获得催化性能更好的Ru/MOF催化剂.鉴于Ru-B/MIL-53(Al)和Ru-B/MIL-100(Fe)催化剂的催化性能差异最为显著,在下文中我们对它们进行了表征,以期为理解Ru-B/MOF催化剂的构效关系提供线索.
3.2 MIL-53(Al)、MIL-100(Fe)载体及催化剂的表征
表2列出了MIL-53(Al)、MIL-100(Fe)载体及相应催化剂的物理化学性质.由表可见,Ru-B/MIL-53(Al)和Ru-B/MIL-100(Fe)催化剂中Ru的实际负载量均接近理论负载量,说明浸渍-化学还原法是一种金属定量负载在MOF材料上的有效方法,且两个催化剂中Ru-B的化学组成相近.与载体相比,负载Ru-B后,催化剂的SBET和孔容Vpore有不同程度的降低,Ru-B/MIL-100(Fe)催化剂的降低更为显著,但平均孔径dpore变化不大.CO脉冲吸附结果表明,Ru-B/MIL-53(Al)催化剂的SRu为17.7 m2·g-1,明显高于Ru-B/MIL-100(Fe)催化剂的1.1 m2·g-1.

表1 Ru-B/MOF催化剂上的苯部分加氢结果aTable 1 Results of the partial hydrogenation of benzene over theRu-B/MOF catalystsa

表2 MIL-53(Al)、MIL-100(Fe)载体及其负载的Ru-B催化剂的物理化学性质Table 2 Physicochemical properties of MIL-53(Al),MIL-100(Fe),and the supported Ru-B catalysts
图2为MIL-53(Al)、MIL-100(Fe)载体及相应催化剂的XRD谱.从图2(A)可见,MIL-53(Al)-as的衍射峰较复杂,但经过DMF溶剂热处理后,得到的MIL-53(Al)载体的衍射峰位置及相对强度与文献22吻合得很好,同时MIL-100(Fe)载体的衍射峰位置及相对强度(图2(B))也与文献27相符,证实我们制备的MIL-53(Al)和MIL-100(Fe)的结构与文献一致.负载Ru-B后,Ru-B/MIL-53(Al)和Ru-B/MIL-100(Fe)催化剂的XRD谱与载体相比没有发生变化,说明负载条件温和的浸渍-化学还原法不会破坏MOF载体的结构.在Ru-B/MIL-53(Al)和Ru-B/MIL-100(Fe)催化剂的谱图中,我们没有观察到除载体以外的衍射峰,这可以归因于Ru-B不仅以非晶态形式存在,而且粒径小于XRD的检测限.38
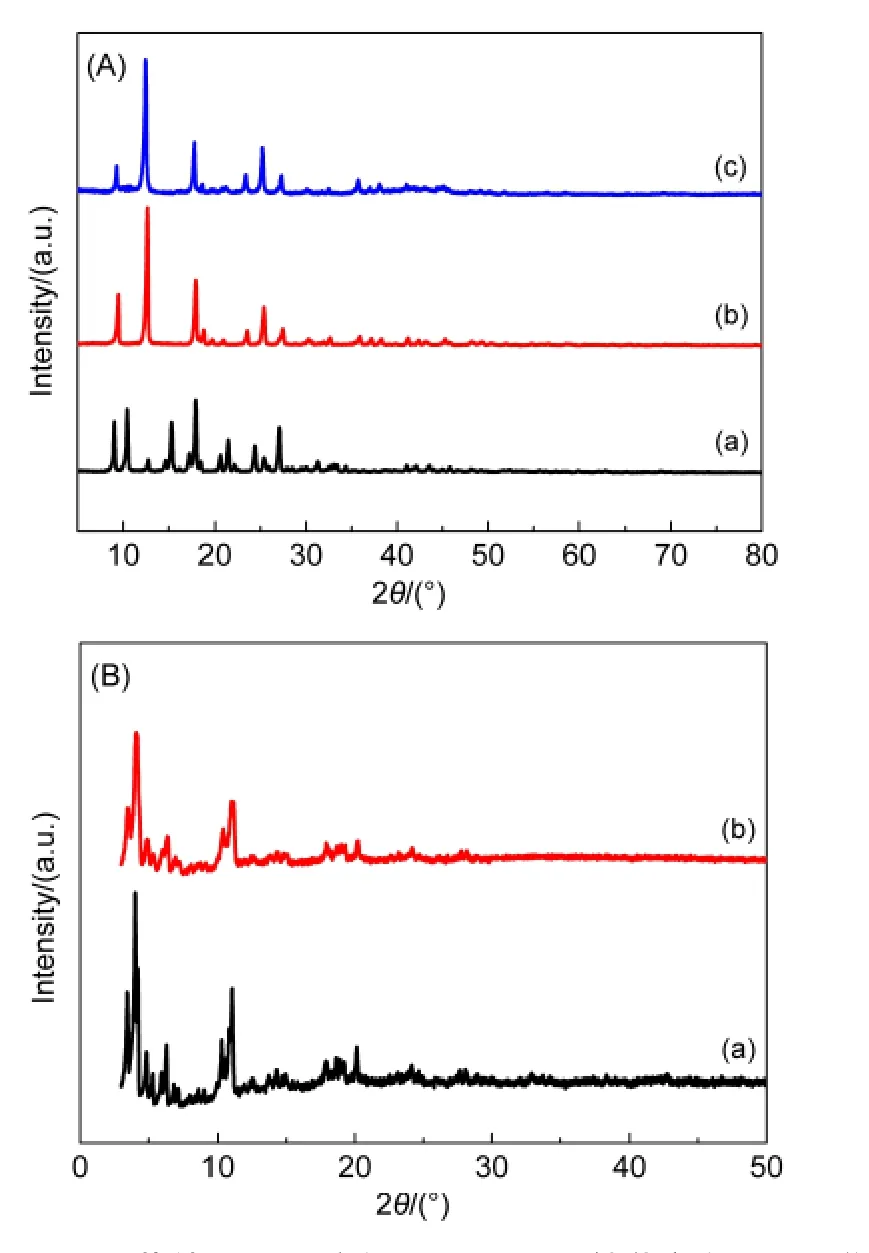
图2 MOF载体以及对应的Ru-B/MOF催化剂的XRD谱Fig.2 XRD patterns of MOF carrier and the corresponding Ru-B/MOF catalysts
图3是H2BDC、MIL-53(Al)载体及Ru-B/MIL-53(Al)催化剂的FT-IR谱.H2BDC在1678和1282 cm-1处出现较强的吸收峰,分别对应于羧基的不对称和对称伸缩振动峰.39对于MIL-53(Al)载体,这两个吸收峰分别移至1579和1413 cm-1,波数差为166 cm-1,明显小于H2BDC的波数差(396 cm-1),表明H2BDC中的羧基与Al3+之间发生了配位作用,13使MOF结构得以形成,与XRD结果一致.负载Ru-B后,Ru-B/MIL-53(Al)催化剂上的羧基不对称和对称伸缩振动峰波数与载体相同,说明Ru-B的负载不破坏MIL-53(Al)载体原有的骨架结构.
MIL-53(Al)载体和Ru-B/MIL-53(Al)催化剂的TGA曲线示于图4.两个样品在74°C前发生轻微失重,归因于样品中物理吸附水或残余有机溶剂的挥发,然后分别在514和435°C时才开始发生明显失重,表明载体和催化剂均有较高的热稳定性.Ru-B/MIL-53(Al)催化剂的失重温度低于MIL-53(Al)载体,可能是因为Ru-B/MIL-53(Al)催化剂中的羧基与Ru发生相互作用,减弱了其与Al3+之间的配位作用,从而使金属-有机骨架结构易于发生分解.13另一方面,羧基与Ru-B之间存在相互作用,将有助于Ru-B在MIL-53(Al)载体上的锚定.MIL-53(Al)载体失重后的剩余质量百分比为20.7%,与理论值21.9%接近(主要为Al2O3).22Ru-B/MIL-53(Al)催化剂的剩余质量百分比为36.3%.结合Al2O3的剩余质量及Ru-B的化学组成(表2),可算得Ru的负载量为11.7%(w),与电感耦合等离子体原子发射光谱(ICP)测得的实际负载量一致.
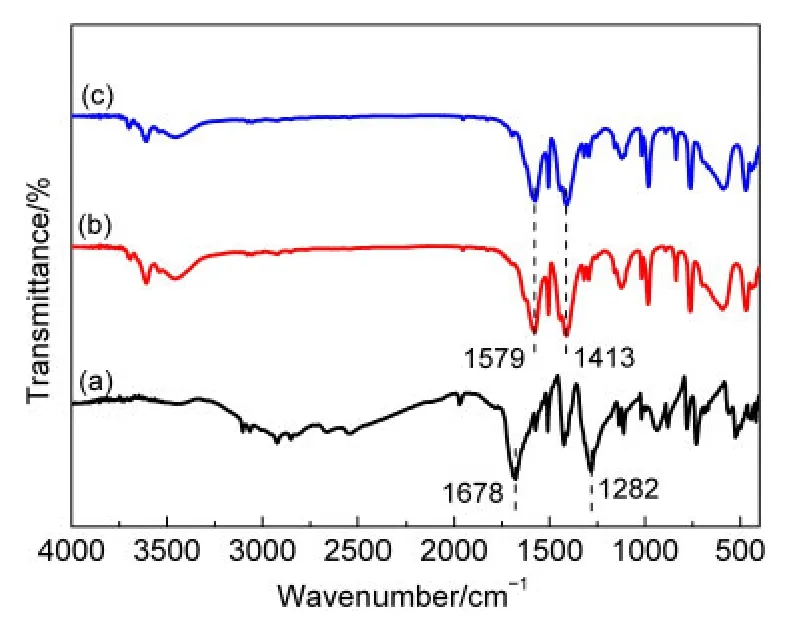
图3 H2BDC(a),MIL-53(Al)载体(b)及Ru-B/MIL-53(Al)催化剂(c)的FTIR谱Fig.3 FTIR spectra of H2BDC(a),MIL-53(Al)carrier(b),and the Ru-B/MIL-53(Al)catalyst(c)

图4 MIL-53(Al)载体(a)和 Ru-B/MIL-53(Al)催化剂(b)的TGA曲线Fig.4 TGAcurves of MIL-53(Al)carrier(a)and the Ru-B/MIL-53(Al)catalyst(b)
图5(A)为Ru-B/MIL-53(Al)催化剂的Ru 3p谱.鉴于Ru最强的Ru 3d峰与C 1s峰发生部分重叠,因此采用Ru 3p峰来确定催化剂中Ru的化学态.由于催化剂中的B含量低(质量百分比约0.7%,相对于载体),加之B的原子灵敏度很低,40故B 1s谱的信噪比差,不能够提供有用的信息.由图5(A)可见,Ru-B/MIL-53(Al)催化剂的Ru 3p1/2和Ru 3p3/2的电子结合能(BE)位于483.6和461.4 eV,与金属Ru一致.41Ru-B/MIL-53(Al)催化剂与Ru箔标样的RuK边X射线吸收近边结构(XANES)谱相似(图5(B)),也支持Ru以金属态存在的结论.
Ru-B/MIL-53(Al)和 Ru-B/MIL-100(Fe)催化剂的形貌见图6.在TEM照片中,Ru-B纳米粒子均匀地分散在MIL-53(Al)载体表面,平均粒径为3.2 nm(图6(a)).Ru-B/MIL-100(Fe)催化剂上的Ru-B纳米粒子则发生严重团聚,平均粒径为46.6 nm(图6(b)),远大于Ru-B/MIL-53(Al)催化剂.结合XRD表征结果,我们推测TEM观察到的Ru-B/MIL-100(Fe)催化剂上的颗粒可能是由更小的初级粒子聚集而成的,因此在图2中不出现衍射峰.图6中两个催化剂的SAED图中均只有弥散的衍射环,而不是分立的衍射环或衍射点,说明Ru-B具有非晶态结构.42
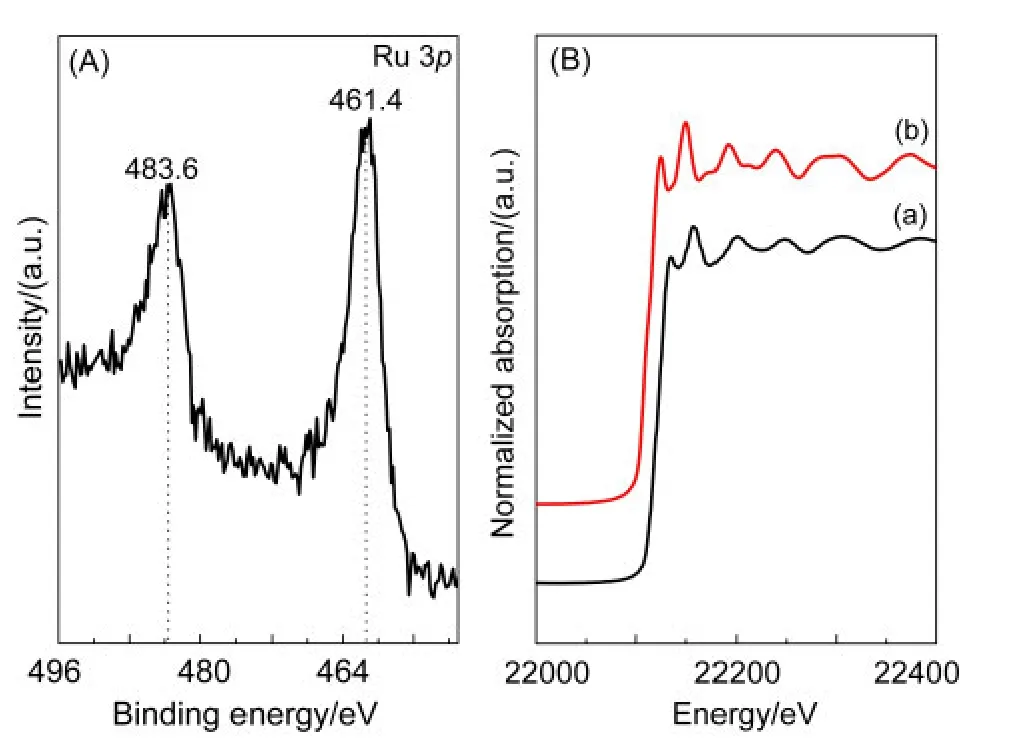
图5 (A)Ru-B/MIL-53(Al)催化剂的Ru 3p XPS谱和(B)Ru-B/MIL-53(Al)催化剂(a)及Ru箔标样(b)的Ru K边XANES谱Fig.5 (A)Ru 3p XPS spectra of the Ru-B/MIL-53(Al)catalyst and(B)normalized Ru K-edge XANES spectra of the Ru-B/MIL-53(Al)catalyst(a)and the Ru foil(b)
由于非晶态合金具有独特的短程有序、长程无序结构,43基于长程有序结构的XRD和SAED方法不能够揭示其结构信息,因此我们采用扩展X射线吸收精细结构(EXAFS)对非晶态Ru-B合金的微观结构进行了表征.图7为Ru-B/MIL-53(Al)催化剂和Ru箔标样经傅里叶变换后的RuK边径向分布函数.在图中,Ru箔除在0.236 nm处出现第一配位层的峰外,还在0.348和0.429 nm处出现第二和第三配位层的峰.但Ru-B/MIL-53(Al)催化剂仅出现第一配位层的峰,符合非晶态合金短程有序但长程无序的结构特征,44有力地证明了催化剂中的Ru-B具有非晶态结构.表3列出了由EXAFS数据拟合得到的Ru-B/MIL-53(Al)催化剂和Ru箔的结构参数.从图8可见拟合得到的k2χ(k)曲线(k2权重的Ru元素K吸收边扩展X射线精细结构)与实验点吻合较好.从表3可见,Ru-B/MIL-53(Al)催化剂中Ru-Ru的Rj为0.271 nm,略大于Ru箔中Ru-Ru的Rj(0.268 nm),这与王晓光等45报道的非晶态Ni-B合金的Ni-Ni配位距离大于Ni箔类似.Ru-B/MIL-53(Al)催化剂的Ru-Ru配位数则明显小于Ru箔,说明在非晶态Ru-B合金中Ru配位高度不饱和.Ru-B/MIL-53(Al)催化剂中还存在Ru-B配位,其N为2.1,Rj为0.218 nm,表明二者之间形成了合金.

图6 催化剂的TEM照片及粒径分布Fig.6 TEM images and particle size distribution histograms of the catalysts
3.3 催化剂构效关系探讨
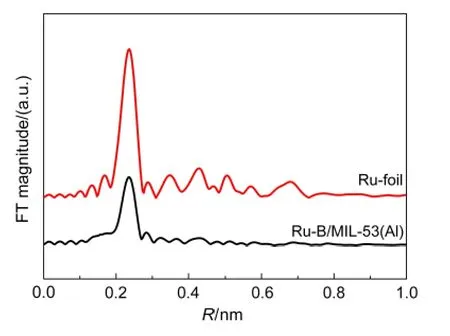
图7 Ru-B/MIL-53(Al)催化剂和Ru箔标样Ru元素K吸收边EXAFS数据经傅里叶变换(FT)得到的径向分布函数Fig.7 Radial distribution functions after Fourier transformation(FT)of the Ru K-edge EXAFS data of the Ru-B/MIL-53(Al)catalyst and the Ru foil
由上述表征可知,Ru-B/MIL-53(Al)和Ru-B/MIL-100(Fe)催化剂上Ru负载量、Ru-B合金的组成接近,且二者均具有非晶态结构,然而在苯部分加氢反应中,前者的催化加氢活性远高于后者.CO脉冲吸附结果表明,Ru-B/MIL-53(Al)催化剂的SRu远大于Ru-B/MIL-100(Fe)催化剂,与TEM观察到前者的Ru-B纳米粒子粒径更小、分散度更高一致.Ru-B/MIL-53(Al)催化剂中高度分散的Ru-B纳米粒子能够提供更多的活性中心,故在苯部分加氢反应中显示出更高的催化活性.由表2可见,虽然MIL-100(Fe)载体的SBET比MIL-53(Al)载体大732 m2·g-1,但大的SBET并没有导致高的Ru-B分散度,这意味着与MOF载体的相互作用对Ru-B纳米粒子的分散有着重要的影响,可以在后续的工作中有目的地加以调控,以改善催化剂的性能.根据SRu和r0,可以算得Ru-B/MIL-53(Al)和Ru-B/MIL-100(Fe)催化剂的苯加氢转换频率(TOF),其值分别为11和3.3 s-1,即前者更高的催化活性不仅是其SRu更大的原因,其本征活性也更高.这可能是Ru-B/MIL-53(Al)催化剂中存在强的金属-载体相互作用,不仅有利于Ru-B纳米粒子的高分散,也提高了Ru-B纳米粒子的活性.另一方面,Ronchin和Toniolo46认为,在液相苯部分加氢反应中,由于生成环己烷比生成环己烯需要的表面氢物种更多,这会要求在同一个纳米粒子上有更多相邻的活性位参与到吸附和反应中来.较大的Ru-B纳米粒子可以提供更多相邻的活性位,有利于环己烷的生成.因此,在Ru-B粒径小的Ru-B/MIL-53(Al)催化剂上,其S0较Ru-B/MIL-100(Fe)催化剂为高.
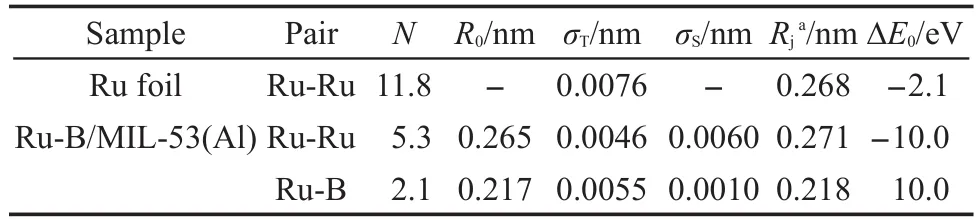
表3 Ru K边EXAFS数据拟合得到的Ru-B/MIL-53(Al)催化剂及Ru箔标样的结构参数Table 3 Structural parameters derived from the Ru K-edge EXAFS data of the Ru-B/MIL-53(Al)catalyst and the Ru foil
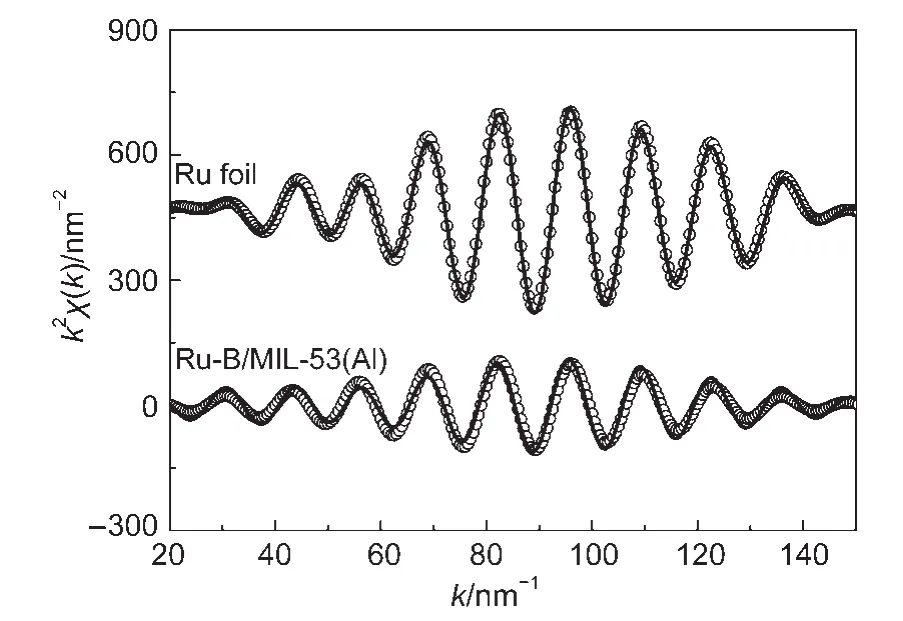
图8 Ru-B/MIL-53(Al)催化剂和Ru箔标样的实验k2χ(k)数据(○)及拟合曲线(—)Fig.8 Experimental k2χ(k)data(○)and fitted curves(—)of the Ru-B/MIL-53(Al)catalyst and the Ru foil
3.4 反应条件的影响

图9 反应温度对Ru-B/MIL-53(Al)催化剂苯部分加氢催化性能的影响Fig.9 Effect of reaction temperature on the catalytic performance of the Ru-B/MIL-53(Al)catalyst in the partial hydrogenation of benzene
图9考察了反应温度对Ru-B/MIL-53(Al)催化剂的苯部分催化加氢性能的影响.在图9(a)中,随着反应温度由140°C提高到160°C,苯加氢速率增加.但继续提高至180°C,反应速率又开始下降.在图9(b)中,随着反应温度由140°C提高到180°C,环己烯选择性和得率升高.但继续升温至190°C,选择性及得率反而降低.从环己烯得率上看,Ru-B/MIL-53(Al)催化剂的最佳反应温度为180°C,此时环己烯得率可达24%,高于一些文献中报道的结果.47−49提高反应温度有利于克服反应能垒,加快苯加氢反应的进行.然而,过高的反应温度使H2在液相中的溶解度降低,从而导致苯加氢反应速率下降.14就温度对选择性的影响而言,提高温度一方面有利于环己烯从催化剂表面脱附,避免过度加氢;另一方面使催化剂表面的氢覆盖度降低,抑制环己烯进一步加氢.这两方面的效应均有利于环己烯选择性的提高.但过高的反应温度会增加环己烯在催化剂表面滞水层中的溶解度,提高环己烯在催化剂表面的覆盖度和加氢速率,从而导致选择性下降.50
图10考察了在180°C反应温度下,H2压力对Ru-B/MIL-53(Al)催化剂苯部分加氢催化性能的影响.由图可知,随H2压力由4 MPa提高到6 MPa,苯加氢速率提高,但环己烯选择性和得率先升高,然后降低,最佳H2压力为5 MPa.对在Ru催化剂上的苯部分加氢反应,反应速率对H2压力为1级.50随着H2压力升高,催化剂表面的氢覆盖度变大,因此苯加氢速率变快,同时也有利于环己烯的生成.51然而,由于环己烯的加氢能垒远低于苯,52在更高的H2压力下,环己烯继续加氢生成环己烷变得更加有利,导致环己烯选择性下降.53

图10 H2压力对Ru-B/MIL-53(Al)催化剂苯部分加氢催化性能的影响Fig.10 Effect of H2pressure on the catalytic performance of the Ru-B/MIL-53(Al)catalyst in the partial hydrogenation of benzene
4 结论
采用浸渍-化学还原法在保持MOF材料原有结构的同时,能够方便地将非晶态Ru-B合金纳米粒子负载在MOF材料上.在苯部分加氢反应中,MOF材料的性质对负载型非晶态Ru-B催化剂的催化性能有着显著的影响.在所考察的7种MOF材料中,MIL-53(Al)负载的非晶态Ru-B催化剂显示了最高的苯加氢活性和环己烯选择性,归因于Ru-B纳米粒子在MIL-53(Al)载体上的高度分散.在180°C和5 MPa H2压力下,以乙醇胺为修饰剂,Ru-B/MIL-53(Al)催化剂上的环己烯得率可达24%,证明了MOF材料用作苯部分加氢催化剂载体的可行性.通过调变MOF材料中金属离子的种类、有机配体上的取代基团、孔道尺寸和结构等,Ru-B/MOF催化剂在苯部分加氢反应中的催化性能有望进一步提高.
(1) Rosi,N.L.;Eckert,J.;Eddaoudi,M.;Vodak,D.T.;Kim,J.;O′Keeffe,M.;Yaghi,O.M.Science2003,300,1127.doi:10.1126/science.1083440
(2) Zeng,Y.Y.;Zhang,B.J.Acta Phys.-Chim.Sin.2008,24,1493.[曾余瑶,张秉坚.物理化学学报,2008,24,1493.]doi:10.3866/PKU.WHXB20080828
(3) Li,J.R.;Kuppler,R.J.;Zhou,H.C.Chem.Soc.Rev.2009,38,1477.doi:10.1039/b802426j
(4) Lee,J.;Farha,O.K.;Roberts,J.;Scheidt,K.A.;Nguyen,S.T.;Hupp,J.T.Chem.Soc.Rev.2009,38,1450.doi:10.1039/b807080f
(5) Liang,Q.;Zhao,Z.;Liu,J.;Wei,Y.C.;Jiang,G.Y.;Duan,A.J.Acta Phys.-Chim.Sin.2014,30,129.[粱 倩,赵 震,刘 坚,韦岳山,姜桂元,段爱军.物理化学学报,2014,30,129.]doi:10.3866/PKU.WHXB201311201
(6) Zhou,Y.X.;Liang,S.G.;Song,J.L.;Wu,T.B.;Hu,S.Q.;Liu,H.Z.;Jiang,T.;Han,B.X.Acta Phys.-Chim.Sin.2010,26,939.[周印羲,粱曙光,宋金良,吴天斌,胡素琴,刘会贞,姜 涛,韩布兴.物理化学学报,2010,26,939.]doi:10.3866/PKU.WHXB20100406
(7) Liu,Y.;Mo,K.;Cui,Y.Inorg.Chem.2013,52,10286.doi:10.1021/ic400598x
(8) Gu,X.;Lu,Z.H.;Jiang,H.L.;Akita,T.;Xu,Q.J.Am.Chem.Soc.2011,133,11822.doi:10.1021/ja200122f
(9) Uemura,T.;Kitaura,R.;Ohta,Y.;Nagaoka,M.;Kitagawa,S.Angew.Chem.Int.Edit.2006,45,4112.
(10) Jiang,H.L.;Akita,T.;Ishida,T.;Haruta,M.;Xu,Q.J.Am.Chem.Soc.2011,133,1304.doi:10.1021/ja1099006
(11) Proch,S.;Herrmannsdorfer,J.;Kempe,R.;Kern,C.;Jess,A.;Seyfarth,L.;Senker,J.Chem.-Eur.J.2008,14,8204.doi:10.1002/chem.v14:27
(12) Schröder,F.;Esken,D.;Cokoja,M.;van den Berg,M.W.E.;Lebedev,O.I.;Van Tendeloo,G.;Walaszek,B.;Buntkowsky,G.;Limbach,H.H.;Chaudret,B.;Fischer,R.A.J.Am.Chem.Soc.2008,130,6119.doi:10.1021/ja078231u
(13)Wu,T.B.;Zhang,P.;Ma,J.;Fan,H.L.;Wang,W.T.;Jiang,T.;Han,B.X.Chin.J.Catal.2013,34,167.doi:10.1016/S1872-2067(11)60475-0
(14)Wang,W.T.;Liu,H.Z.;Ding,G.D.;Zhang,P.;Wu,T.B.;Jiang,T.;Han,B.X.ChemCatChem2012,4,1836.doi:10.1002/cctc.v4.11
(15) Odenbrand,C.U.I.;Andersson,S.L.T.J.Chem.Technol.Biotechnol.1982,32,365.
(16)Wang,J.Q.;Wang,Y.Z.;Xie,S.H.;Qiao,M.H.;Li,H.X.;Fan,K.N.Appl.Catal.A2004,272,29.doi:10.1016/j.apcata.2004.04.038
(17) Sun,H.J.;Jiang,H.B.;Li,S.H.;Wang,H.X.;Pan,Y.J.;Dong,Y.Y.;Liu,S.C.;Liu,Z.Y.Chin.J.Catal.2013,34,684.doi:10.1016/S1872-2067(11)60489-0
(18) Liu,J.L.;Zhu,Y.;Liu,J.;Pei,Y.;Li,Z.H.;Li,H.;Li,H.X.;Qiao,M.H.;Fan,K.N.J.Catal.2009,268,100.doi:10.1016/j.jcat.2009.09.007
(19) Nagahara,H.;Ono,M.;Konishi,M.;Fukuoka,Y.Appl.Surf.Sci.1997,121-122,448.
(20) Millange,F.;Serre,C.;Guillou,N.;Férey,G.;Walton,R.I.Angew.Chem.Int.Edit.2008,47,4100.
(21) Serre,C.;Millange,F.;Thouvenot,C.;Noguès,M.;Marsolier,G.;Loue¨r,D.;Férey,G.J.Am.Chem.Soc.2002,124,13519.doi:10.1021/ja0276974
(22) Loiseau,T.;Serre,C.;Huguenard,C.;Fink,G.;Taulelle,F.;Henry,M.;Bataille,T.;Férey,G.Chem.-Eur.J.2004,10,1373.
(23) Férey,G.;Millange,F.;Morcrette,M.;Serre,C.;Doublet,M.L.;Grenèche,J.M.;Tarascon,J.M.Angew.Chem.Int.Edit.2007,46,3259.
(24) Cavka,J.H.;Jakobsen,S.;Olsbye,U.;Guillou,N.;Lamberti,C.;Bordiga,S.;Lillerud,K.P.J.Am.Chem.Soc.2008,130,13850.doi:10.1021/ja8057953
(25) Trung,T.K.;Trens,P.;Tanchoux,N.;Bourrelly,S.;Llewellyn,P.L.;Loera-Serna,S.;Serre,C.;Loiseau,T.;Fajula,F.;Férey,G.J.Am.Chem.Soc.2008,130,16926.doi:10.1021/ja8039579
(26) Férey,G.;Mellot-Draznieks,C.;Serre,C.;Millange,F.;Dutour,J.;Surblé,S.;Margiolaki,I.Science2005,309,2040.doi:10.1126/science.1116275
(27)Yoon,J.W.;Seo,Y.K.;Hwang,Y.K.;Chang,J.S.;Leclerc,H.;Wuttke,S.;Bazin,P.;Vimont,A.;Daturi,M.;Bloch,E.Angew.Chem.Int.Edit.2010,49,5949.doi:10.1002/anie.201001230
(28) Valenzano,L.;Civalleri,B.;Chavan,S.;Bordiga,S.;Nilsen,M.H.;Jakobsen,S.;Lillerud,K.P.;Lamberti,C.Chem.Mater.2011,23,1700.doi:10.1021/cm1022882
(29)Ahnfeldt,T.;Gunzelmann,D.;Loiseau,T.;Hirsemann,D.;Senker,J.;Férey,G.;Stock,N.Inorg.Chem.2009,48,3057.doi:10.1021/ic8023265
(30) Vermoortele,F.;Ameloot,R.;Vimont,A.;Serre,C.;De Vos,D.Chem.Commun.2011,47,1521.doi:10.1039/c0cc03038d
(31)Karim,A.M.;Prasad,V.;Mpourmpakis,G.;Lonergan,W.W.;Frenkel,A.I.;Chen,J.G.;Vlachos,D.G.J.Am.Chem.Soc.2009,131,12230.doi:10.1021/ja902587k
(32) Zhou,G.B.;Liu,J.L.;Tan,X.H.;Pei,Y.;Qiao,M.H.;Fan,K.N.;Zong,B.N.Ind.Eng.Chem.Res.2012,51,12205.
(33) Lengeler,B.;Eisenberger,P.Phys.Rev.B1980,21,4507.doi:10.1103/PhysRevB.21.4507
(34) De Crescenzi,M.;Balsatori,A.;Comin,F.;Incoccia,L.;Mobilio,S.;Motta,N.Solid State Commun.1981,37,921.doi:10.1016/0038-1098(81)91187-X
(35)Ankudinov,A.;Ravel,B.;Rehr,J.J.FEFF8,Version 8.20;University of Washington:Seattle,WA,2002.
(36) Struijk,J.;Moene,R.;van der Kamp,T.;Scholten,J.J.F.Appl.Catal.A1992,89,77.doi:10.1016/0926-860X(92)80079-R
(37) Schwab,F.;Lucas,M.;Claus,P.Angew.Chem.Int.Edit.2011,50,10453.doi:10.1002/anie.201104959
(38) Liu,J.L.;Zhu,L.J.;Pei,Y.;Zhuang,J.H.;Li,H.;Li,H.X.;Qiao,M.H.;Fan,K.N.Appl.Catal.A2009,353,282.doi:10.1016/j.apcata.2008.10.056
(39) Zhao,Y.J.;Zhang,J.L.;Han,B.X.;Song,J.L.;Li,J.S.;Wang,Q.Angew.Chem.Int.Edit.2011,50,636.doi:10.1002/anie.v50.3
(40) Moulder,J.F.;Stickle,W.F.;Sobol,P.E.;Bomben,K.D.Handbook of X-ray Photoelectron Spectroscopy;Chastain,J.Ed.;Perkin-Elmer:Eden Prairie,Minnesota,1992;p 253.
(41) Campbell,P.S.;Santini,C.C.;Bayard,F.;Chauvin,Y.;Collière,V.;Podgoraek,A.;Costa Gomes,M.F.;Sá,J.J.Catal.2010,275,99.doi:10.1016/j.jcat.2010.07.018
(42)Xie,S.H.;Qiao,M.H.;Li,H.X.;Wang,W.J.;Deng,J.F.Appl.Catal.A1999,176,129.doi:10.1016/S0926-860X(98)00232-4
(43) Pei,Y.;Zhou,G.B.;Luan,N.;Zong,B.N.;Qiao,M.H.;Tao,F.Chem.Soc.Rev.2012,41,8140.doi:10.1039/c2cs35182j
(44) Pei,Y.;Guo,P.J.;Qiao,M.H.;Li,H.X.;Wei,S.Q.;He,H.Y.;Fan,K.N.J.Catal.2007,248,303.doi:10.1016/j.jcat.2007.03.024
(45)Wang,X.G.;Yan,W.S.;Zhong,W.J.;Zhang,X.Y.;Wei,S.Q.Chem.J.Chin.Univ.2001,22,349.[王晓光,闫文胜,钟文杰,张新夷,韦世强.高等学校化学学报,2001,22,349.]
(46) Ronchin,L.;Toniolo,L.Catal.Today1999,48,255.doi:10.1016/S0920-5861(98)00380-0
(47) Hronec,M.;Cvengroaová,Z.;Králik,M.;Palma,G.;Corain,B.J.Mol.Catal.A1996,105,25.doi:10.1016/1381-1169(95)00184-0
(48) Mazzieri,V.A.;L′Argentiére,P.C.;Fígoli,N.S.React.Kinet.Catal.Lett.2004,81,107.doi:10.1023/B:REAC.0000016523.15129.90
(49) Silveira,E.T.;Umpierre,A.P.;Rossi,L.M.;Machado,G.;Morais,J.;Soares,G.V.;Baumvol,I.J.R.;Teixeira,S.R.;Fichtner,P.F.P.;Dupont,J.Chem.-Eur.J.2004,10,3734.
(50)Struijk,J.;d′Angremond,M.;Lucas-de Regt,W.J.M.;Scholten,J.J.F.Appl.Catal.A1992,83,263.doi:10.1016/0926-860X(92)85039-E
(51) Ronchin,L.;Toniolo,L.Appl.Catal.A2001,208,77.doi:10.1016/S0926-860X(00)00690-6
(52) Fan,C.;Zhu,Y.A.;Zhou,X.G.;Liu,Z.P.Catal.Today2011,160,234.doi:10.1016/j.cattod.2010.03.075
(53) Liu,H.Z.;Liang,S.G.;Wang,W.T.;Jiang,T.;Han,B.X.J.Mol.Catal.A2011,341,35.doi:10.1016/j.molcata.2011.03.021
猜你喜欢
化工生产与技术(2024年1期)2024-05-31 13:54:58
时代英语·高一(2024年3期)2024-04-29 00:00:00
机械制造(2020年5期)2020-02-20 03:41:19
第一财经(2019年8期)2019-08-26 17:53:46
物理学报(2017年17期)2017-09-09 01:01:36
中国科技纵横(2016年19期)2016-12-10 13:58:08
哈尔滨医药(2015年2期)2015-12-01 03:57:13
学习月刊(2015年14期)2015-07-09 03:37:48
山西大同大学学报(自然科学版)(2015年1期)2015-01-22 07:14:12
中国造纸(2014年1期)2014-03-01 02:10:08
