
石莼多糖的单糖组成成分分析
2014-05-10 06:05冯学珍陈颖伍善广
食品工业科技 2014年7期
冯学珍,陈颖,伍善广*
(1.广西科技大学,广西 柳州545006;2.广东食品药品职业学院,广东 广州,510520)
海藻多糖是海洋生物多糖中种类繁多、资源较丰富的一类,其广泛的生物活性使人们对海藻多糖的研究越来越重视,成为目前海洋药物研究中较为活跃的领域之一[1-2]。石莼为绿藻石莼科植物石莼(Ulva lactuca L.)的干燥藻体,亦称海白菜、海青菜、海莴苣等,广泛分布在太平洋沿海,资源十分丰富。《本草纲目》中记载石莼具有软坚散结、清热解毒、利水消肿等功效[3]。现代药理学研究表明,石莼多糖具有抗氧化、抗菌、抗病毒和降血糖等作用[3-5],但关于石莼多糖的单糖组成却鲜有报道,石莼由于采集区域、采摘时期的不同,可能造成单糖组成的迥异,因此研究和改进石莼单糖组分的测定方法为进行多糖质量控制和获取多糖基本信息提供科学依据。
目前多糖的单糖组成分析方法主要有薄层色谱法(TLC)、气相色谱法(GC)和高效液相色谱法(HPLC)等[6]。其中柱前衍生化 HPLC法因其操作简单,分离效果好,重现性强等优点已成为检测多糖中单糖组成较常用的方法[6-8]。糖类衍生的试剂 1-苯基-3-甲基-5-吡唑酮(PMP)可以在温和的反应条件下与糖醛基进行定量反应,生成单糖-PMP衍生物,该衍生物不易产生异构峰并在250 nm波长处有强吸收,因此常常用PMP作为衍生化试剂用于多糖化合物的单糖组成分析[6-8]。本研究分别采用薄层色谱法和PMP柱前衍生化HPLC法对超声提取的石莼多糖的单糖组成进行了分析,并对两种方法进行了比较,为该多糖的进一步开发利用提供参考。
1 材料与方法
1.1 材料与仪器
石莼,采自广西北部湾海域,经大连海洋大学邢坤副教授鉴定为绿藻门石莼属黑石礁石莼( Ulva lactuca L)的新鲜藻体。反复水洗几次,以除去泥沙和盐分等杂质,蒸馏水洗2次,干燥、粉碎、过筛后置于干燥器中备用。
葡萄糖(Glc)、甘露糖(Man)、半乳糖(Gal)、鼠李糖(Rha)、木糖(Xyl)、核糖(Rib)标准品,均购自国药集团化学试剂有限公司;硅胶G(分析纯),购自青岛海洋化工厂;甲醇、乙腈均为色谱纯,1-苯基-3-甲基-5-吡唑酮(PMP,99.0%)、三氟乙酸( TFA)、乙酸铵、氯仿、氢氧化钠等均为分析纯,水为超纯水。
Agilent 1260型高效液相色谱仪,美国Agilent 公司(包括 G1311C型四元泵、G1314B型VWD检测器、AXW-8型柱温箱等);Diamonsil C18色谱柱 (250mm×4.6mm,5μm), 迪马公司;TU-1901双光束紫外可见分光光度计, 北京普析通用仪器有限责任公司;PHS-25 酸度计,上海虹益仪器仪表有限公司;XW-80A漩涡混合器, 上海精科实业有限公司等。
1.2 石莼多糖的提取
称取100g石莼干燥粉末,95%乙醇浸泡过夜,滤渣风干后加50倍水,于70℃超声波提取40min。将提取液离心,滤渣重复提取2次,合并提取液进行减压浓缩。浓缩液加95%乙醇沉淀2次,4℃过夜后离心干燥即得粗多糖。
将得到的粗多糖采用酶法脱蛋白3次[9]:1.0%多糖溶液加1.0%的胃蛋白酶在50℃、pH6.5条件下水解 2h,沸水浴灭酶 5min,冷却后离心,取上清液加 95%乙醇沉淀,4℃过夜后离心干燥即得石莼多糖样品。将样品在波长260~280nm范围内扫描,结果显示无蛋白吸收峰时即表明蛋白已经除尽。
1.3 石莼多糖的水解[8]
精密称取30mg石莼多糖样品于具塞试管中,加入2mol/L的三氟乙酸(TFA)2mL,封管后于110℃水解2h,冷却至室温离心,上清液用NaOH调pH值至7.0,定容至5mL即得石莼多糖样品水解液[8]。
1.4 薄层色谱法
精密称取Glc、 Man、Gal、Rha、Xyl、Rib的单糖标准品,加水制成1mg/mL的水溶液。
取单糖标准品溶液和石莼多糖的样品水解液,点于硅胶薄层板上,以正丁醇-丙酮-水(4:3:1)为展开剂进行展开,取出,吹干后,喷显色剂苯胺-邻苯二甲酸(邻苯二甲酸1.66g溶于水饱和的正丁醇100ml,加苯胺0.93g),置于105℃烘箱内,显色8min。
1.5 柱前衍生 HPLC法
1.5.1 石莼多糖样品水解液的衍生[10]
准确吸取100μL样品水解液于离心管中,加入100μL 0.5mol/L PMP甲醇溶液及100μL 0.3mol/L NaOH溶液,涡旋混合后在70℃水浴中反应30min[10],取出冷却,加100μL 0.3mol/L HCl溶液中和,再加水1mL、氯仿1mL进行萃取,弃去氯仿层,共萃取 3 次,合并水层,水层用 0.45 μm 微孔膜过滤后供 HPLC 进样分析。
1.5.2 单糖标准品衍生物的制备
取各种单糖(Glc、Man、Gal、Rha、Xyl、Rib)标准品用超纯水配成0.05mol/L水溶液,得到6种单糖的标准溶液,各取相同体积的6种单糖标准溶液即得单糖混合标准溶液。分别取6种单糖标准溶液及单糖混合标准溶液各100μL,按照“1.5.1”项下方法进行衍生化处理,0.45μm 微孔膜过滤后供 HPLC 进样分析。
1.5.3 色谱条件
色谱柱:Diamonsil C18(250mm×4.6mm, 5μm);流动相:A相为0.05mol/L乙酸铵缓冲溶液(pH 5. 5),B相为乙腈;洗脱梯度(0 min, 0%B; 10min, 18%B; 25min,25%B;30min,30%B);柱温:30℃;流速 1.0mL/min;检测波长:250nm。
1.5.4 方法学考察
取不同量的PMP衍生单糖标准溶液,在“1.5.3”色谱条件下进样分析,根据测定的峰面积对相应单糖的浓度进行数据分析即得单糖的回归方程。再将PMP 衍生的单糖标准溶液不断稀释,依次进样分析,计算信噪比为3时所对应的6种单糖标准溶液的质量浓度以确定最低检测限(LOD)。
进样吸取单糖混合标准衍生样品溶液10 μL在“1.5.3”色谱条件下重复进样5次,分析6种单糖各色谱峰保留时间和峰面积值的一致性,以考察精密度。
方法重复性试验 按照“1.5”项下的方法平行制备5份石莼多糖的供试品溶液,在“1.5.3”色谱条件下进样分析,考察供试品的重复性。
称取石莼多糖样品 5份于具塞试管中,加入定量单糖标准品溶液,按“1.3”“1.5.1”项下的方法进行水解和衍生化操作,在“1.5.3”色谱条件下进行色谱分析,记录各色谱峰的峰面积,代入单糖的回归方程,计算回收率,考察回收率。
将单糖混合标准衍生样品溶液及石莼多糖的供试品溶液于冰箱4℃储存,分别在衍生化后0、2、4、6、8、10、12h进样,以稳定性考察。
1.6 数据处理方法
利用Origin8.0对所得数据进行统计处理。
2.结果
2.1 薄层色谱分析
采用薄层色谱测定石莼多糖的单糖组成结果见图 1。根据各单糖标准品的 Rf值,可以初步确定石莼多糖由鼠李糖、木糖、半乳糖、核糖、葡萄糖和甘露糖组成,但由于个别单糖的显色斑点有重叠,因此难以分辨石莼多糖中可能含有的其它单糖。此法虽然操作简单,快速,但由于多糖水解后生成的各单糖其极性比较相近,分离效果不理想。由于薄层色谱分辨率的限制,在多糖样品中含量少的斑点出现模糊和重叠,同时该方法难以进行定量测定。

图1 薄层色谱法测定结果1.核糖2.葡萄糖3.半乳糖4.甘露糖5.鼠李糖6.木糖7.石莼多糖样品Fig. 1 The results of thin layer chromatography1.Rib 2.Glc 3.Gal 4.Man 5.Rha 6.Xyl 7. samples
2.2 衍生化条件的优化
本实验主要考察了衍生化的温度和时间对衍生产物峰面积的影响,其结果如图所示。由图2可以看出随着衍生温度(30、50、70、90、100℃)的升高,所得产物峰面积逐渐增大,当衍生温度超过70℃时,产物峰面积变化不大,因此选择衍生温度为70℃。在70℃水浴中分别衍生0.5、1、2h,由图3可知,产物峰面积随着衍生时间的延长略有增加,但是考虑到衍生时间的延长会相应增多副产物,因此综合考虑出峰时间和峰面积选择衍生时间为0.5 h。

图2 反应温度对衍生化反应的影响Fig.2 Effect of reaction temperature on derivatization

图3 反应时间对衍生化反应的影响Fig.3 Effect of reaction time on derivatization
2.3 单糖标准曲线
以质量(y)为横坐标,峰面积(A)为纵坐标绘制标准曲线,即得 6种单糖的线性回归方程,结果见表1。由表1可见,所测定的 6种单糖的峰面积与进样量之间呈良好线性关系。将 PMP 衍生的单糖标准溶液不断稀释进样,计算当信噪比为 3 时所对应的标准溶液的质量以确定最低检测限(LOD),结果见表 1。结果表明, Man、Rha、Gal的 LOD 为 0.00018μg ,Xyl、Rib 的 LOD 为 0.00015μg,Glc的 LOD 为 0.00019μg。
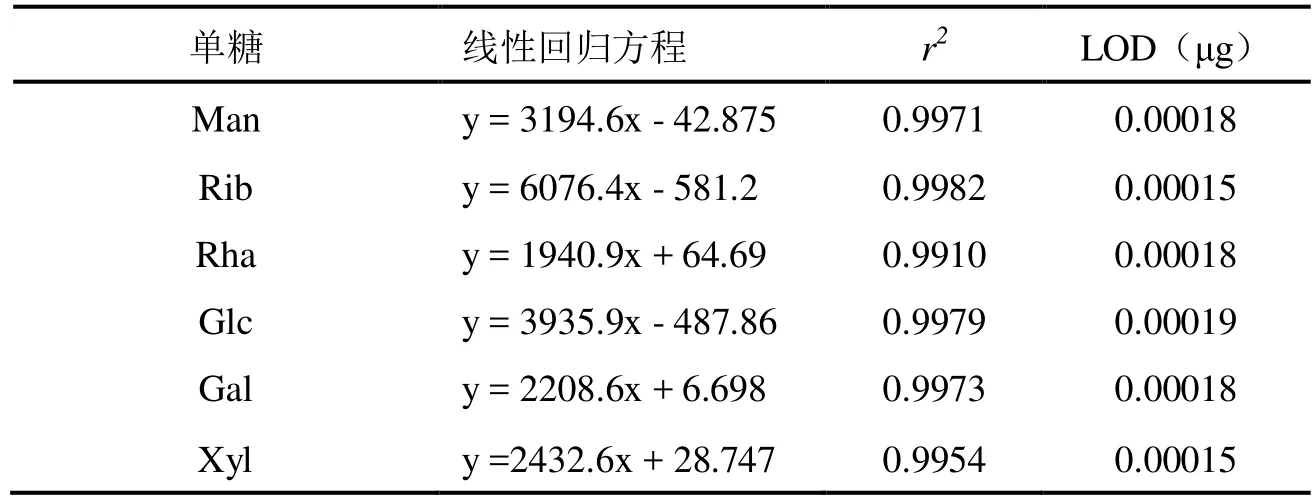
表1 6 种单糖的标准曲线Table.1 Calibration curves of six monosaccharides
2.4 方法学考察结果
2.4.1 精密度试验
结果表明各单糖色谱峰保留时间的相对标准偏差RSD(n=5)为 0.07% ~0.13%;各单糖峰面积的RSD(n=5)为 0.38% ~0.95%,表明精密度良好。
2.4.2 重复性试验
结果表明衍生后石莼多糖中各色谱峰保留时间的RSD(n=5)为 0.69% ~2.25%;各色谱峰峰面积的RSD(n=5)为 2.06% ~4.15%,表明重复性较好。
2.4.3 回收率试验
结果如表2所示,各单糖的平均回收率为 93.78% ~97.42%,各单糖的RSD(n=5)均小于2.73。
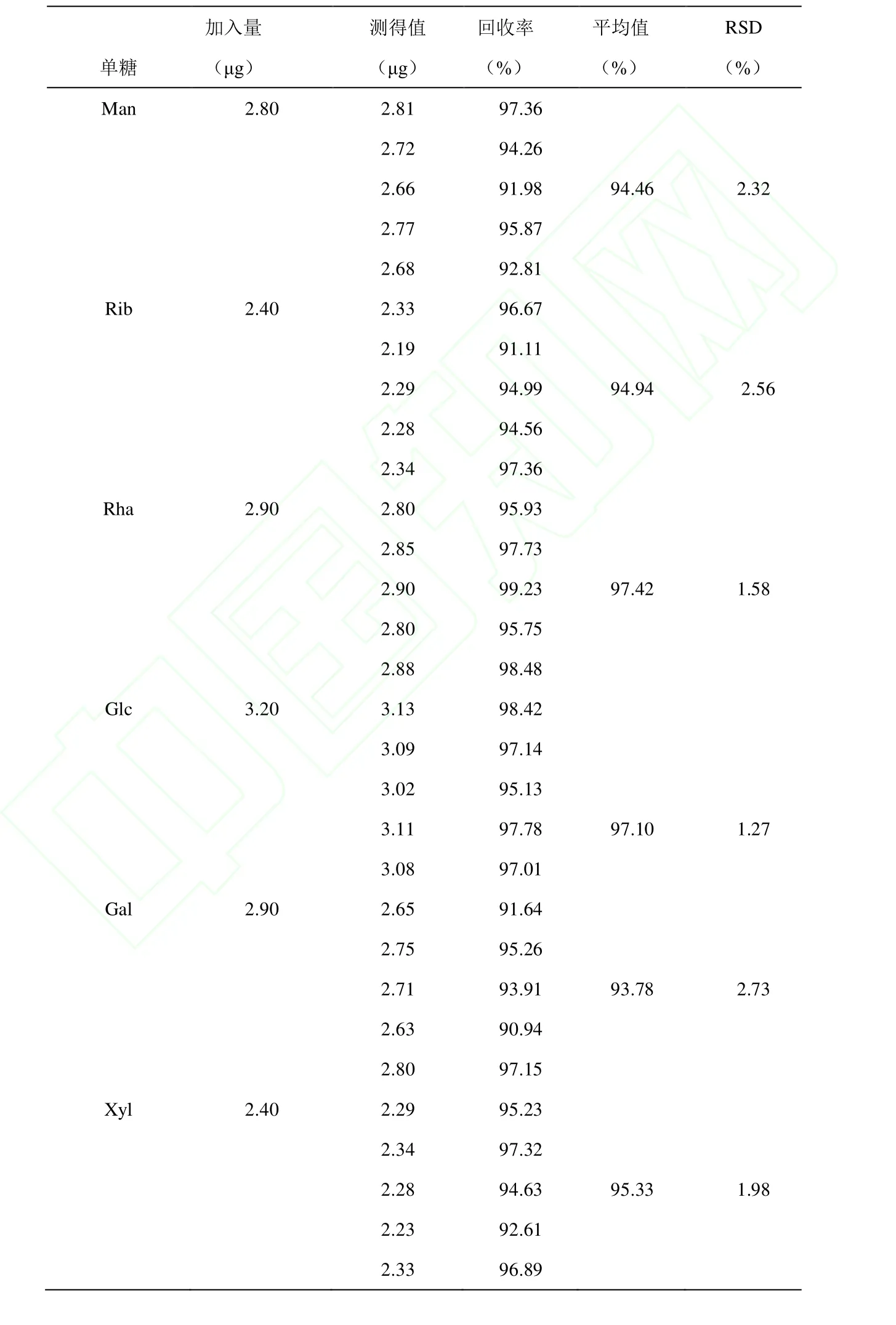
表2 各单糖回收率试验Table.2 Recovery test of six monosaccharides
2.4.4 稳定性试验
结果表明单糖混合标准样品的各色谱峰保留时间和峰面积值的RSD(n=5)均小于2.0%,石莼多糖供试品的各色谱峰保留时间和峰面积值的 RSD(n=5)均小于 5.0%,表明样品在衍生后12小时内稳定性较好。
上述结果表明,用柱前衍生HPLC法分析石莼多糖的单糖组成,操作简便,灵敏度高,准确可靠,重复性好,可以用于石莼多糖的单糖组成测定。
2.5 PMP柱前衍生化HPLC法分析石莼多糖组成
称取石莼多糖 3份,按“1.5”项下的方法进行水解和衍生化操作,在“1.5.3”色谱条件下进行色谱分析。将单糖混合标准品图谱与石莼多糖的图谱对照,色谱图见图4。由图4可见,衍生化后的杂质峰出现在 12min之前,可与各个单糖衍生物的峰很好地分离,不影响单糖成分的分析。石莼多糖的单糖组成包括 Man、Rib、Rha、Glc、Gal 和 Xyl 6种单糖,在 30 min 内均达到基线分离,根据分析和计算,求得石莼多糖中Man、Rib、Rha、Glc、Gal 、Xyl之间的摩尔比为:0.08:0.08:1.00:0.97:0.04:0.68。
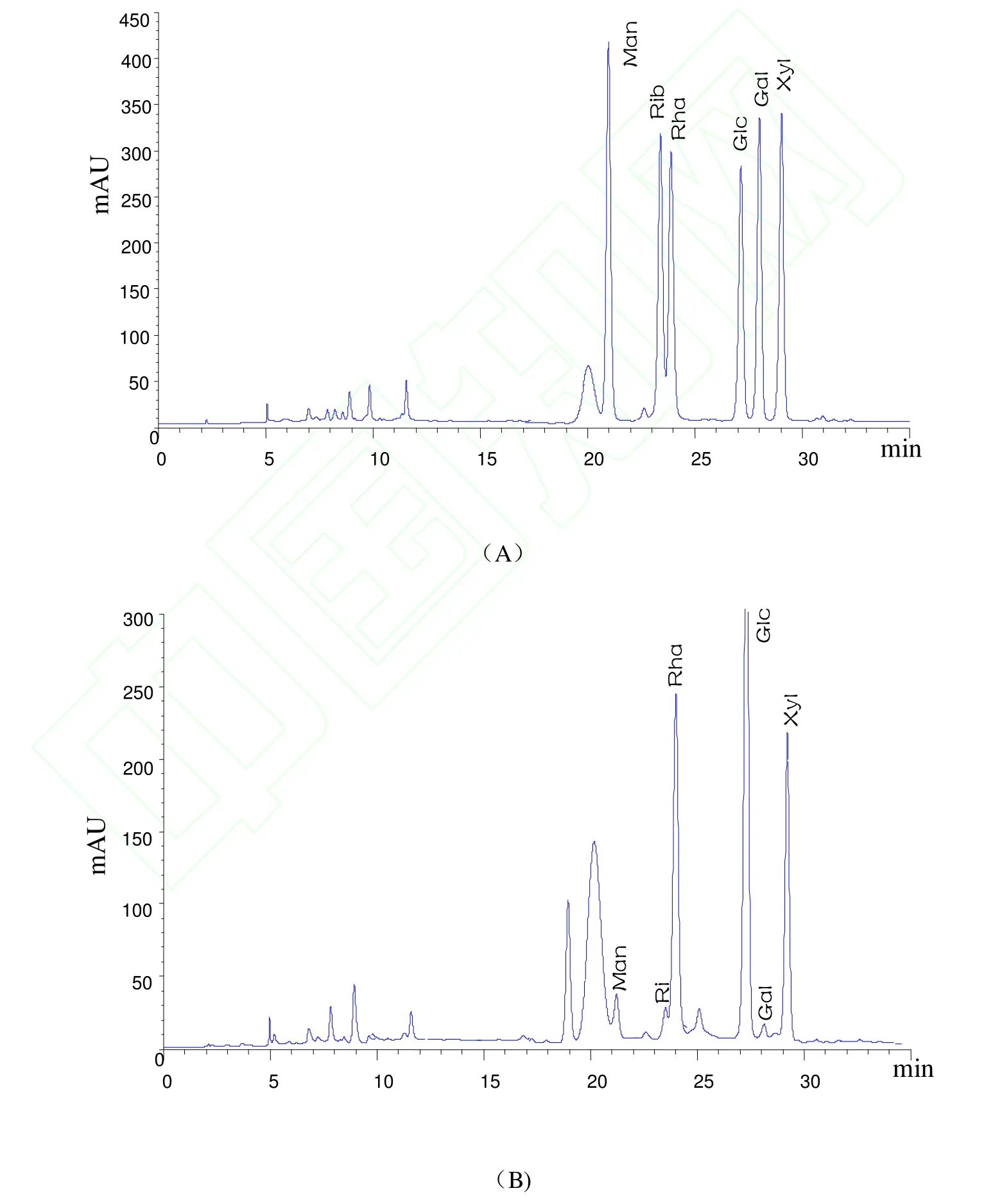
图4 高效液相色谱图Fig.4 HPLC chromatograms of sample
3.讨论与结论
海藻多糖的单糖组成分析常受到自身所含的脂肪、叶绿素、蛋白等杂质的影响,因此多糖的提取分离是成分分析的基础。本研究采用超声波辅助提取、乙醇沉淀、酶法脱蛋白对石莼多糖进行提取和分离。文献[11]研究结果表明超声波提取可提高工作效率,缩短提取时间,节省溶剂等优点,因此本文采用超声辅助提取了石莼多糖,缩短了提取时间,提高了提取效率。
采用薄层色谱法和PMP柱前衍生化HPLC法两种方法分析超声提取的石莼多糖的单糖组成,薄层色谱法结果显示石莼多糖的单糖组成初步推断为鼠李糖、木糖、半乳糖、核糖、葡萄糖和甘露糖组成。PMP柱前衍生化HPLC法结果显示石莼多糖主要由鼠李糖、葡萄糖和木糖组成,此外还含有少量的甘露糖、核糖和半乳糖,各单糖 Man、Rib、Rha、Glc、Gal 、Xyl之间的摩尔比为:0.08:0.08:1.00:0.97:0.04:0.68。TLC 法虽无需衍生化处理,操作简单,但灵敏度低,各单糖极性相近,分离效果差,因此适合于单糖组成较为简单的多糖的定性分析,难以进行定量测定。本文建立的超声辅助提取—柱前衍生HPLC法分析石莼多糖单糖组成的方法操作简单,准确可靠,重复性好,灵敏度高,取样量少,分离效果较理想,适用于石莼多糖样品的单糖组成测定,对进行多糖的化学结构和生物活性的研究具有重要价值,也可用于其它海藻多糖的单糖成分分析。
[1]曾洋洋,韩章润,杨玫婷,等. 海洋糖类药物研究进展[J].中国海洋药物,2013,32(2): 67-72.
[2]韩玲,张淑平,刘晓慧.海藻生物活性物质应用研究进展[J].化工进展,2012,31(8): 1794-1795.
[3]吴淳涛,童国忠,章卢超.石莼提取物抗氧化及抗菌活性研究[J].中国民族民间医药,2011,(6):33-35.
[4]林龙,常建波,孙煜煊.孔石莼多糖降血糖作用研究[J]. 食品科技,2012,37(6):224-227.
[5]张宸阁,高雯欣,卢卫红. 影响辐射小鼠肝脏 SOD 及 MDA 活性的孔石莼多糖研究[J].中医药信息,2012,29(1):111-114.
[6]韩威,姜瑞芝,陈英红,等.银耳多糖单糖组成分析的三种色谱方法比较[J].天然产物研究与开发,2012,24:359-361.
[7]洪碧红,黄文文,尤小云,等. 柱前衍生化高效液相色谱法测定 2-去氧葡萄糖的含量[J]. 中国海洋药物,2011,30(5):44-47.
[8]刘刚,王辉,周本宏,等.高效液相色谱法分析松茸多糖的单糖组成[J].中国医院药学杂志,2012,32(10):758-761.
[9]张岩,马丽娜,王鹏,等. 苦豆子多糖脱蛋白工艺比较[J].中国新药杂志,2012, 21(8):921-925.
[10]郝蕾蕾,张典瑞,赵忠熙.柱前衍生化 HPLC 法测定黄河滩枣多糖的单糖组成[J].中国生化药物杂志,2012,33 (6):740-743.
[11]谌素华,王维民,吴丽萍.绿藻石莼粗多糖提取条件的研究[J].食品研究与开发,2008,29(9):68~71.
猜你喜欢
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
天然产物研究与开发(2019年1期)2019-03-01
临床医药文献杂志(电子版)(2017年11期)2017-05-17
天然产物研究与开发(2016年1期)2016-06-05
海峡科技与产业(2016年3期)2016-05-17
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
云南中医学院学报(2015年3期)2015-07-31
河北工业科技(2015年4期)2015-02-27
