
吸附存储-间歇放电法氧化甲苯的反应过程研究
2014-04-28 06:37王沛涛何梦林鲁美娟樊伟龙吴军良叶代启华南理工大学环境与能源学院广东广州50006华南理工大学广东省大气环境与污染控制重点实验室广东广州50006华南理工大学大气污染控制广东高校工程技术研究中心广东广州50006
中国环境科学 2014年12期
王沛涛,何梦林,鲁美娟,黄 荣,樊伟龙,吴军良,2,叶代启,2,3*(.华南理工大学环境与能源学院,广东 广州 50006;2.华南理工大学广东省大气环境与污染控制重点实验室,广东 广州 50006;3.华南理工大学大气污染控制广东高校工程技术研究中心,广东 广州 50006)
吸附存储-间歇放电法氧化甲苯的反应过程研究
王沛涛1,何梦林1,鲁美娟1,黄 荣1,樊伟龙1,吴军良1,2,叶代启1,2,3*(1.华南理工大学环境与能源学院,广东 广州 510006;2.华南理工大学广东省大气环境与污染控制重点实验室,广东 广州 510006;3.华南理工大学大气污染控制广东高校工程技术研究中心,广东 广州 510006)
以 SBA-15为催化剂,对比连续降解法和吸附存储-间歇放电法净化低浓度甲苯的活性,结果表明吸附存储-间歇放电法下甲苯去除率、碳平衡和CO2选择性更高.运用GC-MS分析了2种降解方式催化剂表面中间产物随时间的变化,苯甲醛进一步氧化分解进而打开苯环,是等离子体催化降解甲苯的控制步骤.对比SBA-15、Mn/SBA-15、Ag/SBA-15、AgMn/SBA-15 4种催化剂在吸附存储-间歇放电法下降解甲苯的活性.结果显示Ag和Mn的引入加速了对2-庚烯醇的氧化催化,AgMn/SBA-15表现出最好的碳平衡、CO2选择性.
非热等离子体催化;甲苯;连续降解;吸附储存-间歇放电;中间产物;降解路径
VOCs是重要的大气污染物,是引起区域大气臭氧超标、PM2.5重度污染的关键前体物.随着社会经济发展,我国 VOCs排放逐年上升[1],由VOCs所导致的大气污染问题进一步凸显.非热等离子技术可以在常温常压下反应,并且反应迅速,但其存在能耗较高、降解不完全、产生二次污染物(O3、CO、NOχ等)等问题[2].催化燃烧法净化效率高、副产物少,但需在一定温度下反应才能进行.为了利用二者优势并解决上述问题,学者们做出了大量研究尝试把非热等离子体与催化结合起来,形成了非热等离子体催化技术,可在常温常压下转化 VOCs,有效提高了反应效率,降低了二次污染[3].应用这种技术时,含VOCs的气流通常是连续地通入等离子体催化反应器(即连续降解法).为了进一步提高等离子体催化系统性能,近期学者们提出了一种新的等离子体催化工作方式——吸附存储-间歇放电法,即把 VOCs的净化分为吸附存储和放电销毁两个阶段,先把VOCs吸附存储于具有吸附性能的多孔催化剂上,然后开启等离子体放电将吸附的 VOCs氧化分解[5-9].然而,目前关于等离子体催化的研究,主要集中在提升能量利用效率及污染物的净化效率,关于识别和减少降解中间产物的研究较少,这些有机副产物也会对环境造成污染,某些甚至比原污染物环境毒性更大.对等离子体催化降解过程生成的中间产物进行识别和分析,将进一步了解其降解过程并促进其机理研究,从而更有效地利用该项技术.
本研究选取低浓度甲苯为目标反应物,以SBA-15为载体,采用浸渍法制备Mn/SBA-15、Ag/SBA-15、AgMn/SBA-15催化剂,考察催化剂在连续降解法和吸附存储-间歇放电法中催化降解甲苯的活性,分析降解过程中有机副产物的变化,探明吸附存储-等离子体销毁法的降解过程,揭示等离子体催化降解甲苯过程中的关键物种.
1 实验方法
1.1 催化剂的制备
采用浸渍法制备一系列的催化剂.首先将一定量的C4H6MnO4·4H2O和AgNO3溶解于无水乙醇中,后加入一定量 SBA-15(先锋纳米,中国),常温条件下搅拌 24h,后于 60℃水浴蒸干,然后在120℃的烘箱中烘干12h,马弗炉中400℃焙烧4h.在本实验中,制备的催化剂分别为 5%Mn/ SBA-15、1%Ag/SBA-15、1%Ag5%Mn/SBA-15(wt.%),催化剂压片、过筛至40~60目备用.
1.2 催化剂的活性评价
本实验装置如图 1所示,高压电源为交流高压变电器(50Hz,0~100kV),使用高压探针(1000:1)测量反应器两端的输出电压,数字功率计(YF9901,中国)测量反应器的输入功率.气体发生装置总流量控制在100mL/min,N2、O2比为4:1,甲苯的浓度为21×10-6.等离子体反应器为线筒式介质阻挡反应器,材料为石英玻璃,反应器内径为6.8mm,外径为 7mm,放电间距为 2.5mm,直径1mm的不锈钢棒作为内电极,外绕的铜丝作为外电极,放电区域长度为 45mm.等离子体反应的消耗功率由输入电压与反应器的电流计算而得,能量密度由(1)式计算.
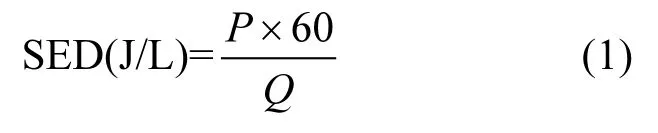
式中:P为反应器的输入功率,W;Q为通入反应器的气体流量,L/min.

图1 实验装置示意Fig.1 Schematic of plasma catalysis for toluene decomposition
在本实验当中,等离子体的有2种工作方式:连续降解法和吸附存储-间歇放电法.如图2所示,在连续降解法下,在放电区域放置 0.2g催化剂,连续通入100mL/min的21×10-6的甲苯同时开启等离子体放电,待反应稳定后测定反应生成的CO与CO2.在吸附存储-间歇放电法下,同样在放电区域中放置0.2g催化剂,吸附存储阶段中通入100mL/min的21×10-6的甲苯72min;在放电降解阶段,停止通入甲苯,通入流量100mL/min 4:1的N2、O2混合气,开启等离子体放电降解,测定反应生成的CO与CO2. 2种降解方式下,等离子体放电的能量密度为均为317J/L.

图2 两种工作方式示意Fig.2 Two kinds of operation mode
反应气体中的甲苯、CO、CO2浓度是由气相色谱(GC-2014C,日本岛津)FID检测器检测,反应器出口的臭氧浓度由臭氧分析仪(IDEAL-1000,山东爱迪尔)测量.甲苯去除效率,碳平衡和CO2选择性是由如式(2)、式(3)、式(4)计算.
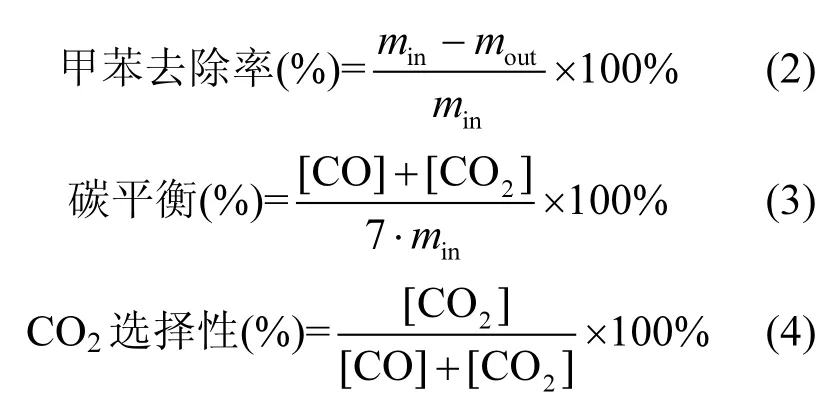
式中:在连续降解法下,min和 mout分别为反应器进、出口单位时间内的甲苯的量(×10-6),[CO]、[CO2]为单位时间内生成的 CO、CO2的量(×10-6);在吸附存储-间歇放电法下,min为吸附存储阶段储存与催化剂中甲苯的总量,mout为放电降解阶段降解反应器出口甲苯的总量(µmol),[CO]、[CO2]为反应产生的CO、CO2的总量(µmol).
催化剂表面的中间产物测定采用溶剂萃取,GC-MS分析.将反应后的催化剂 40mg,加入5mL的CS2(色谱纯,上海阿拉丁),超声萃取30min,上清液用过滤膜(0.22um,中国津腾)过滤,后用GC-MS(GCMS-QP2010Ultra,日本岛津)进行分析.GC-MS分析条件为:色谱柱箱温度为40℃,进样口温度为 200℃,检测器温度为 250℃,分流比10,进样体积为 3mL.设置柱箱程序升温,升温条件为:初始温度40℃停留时间为2min,以6℃/min升温速率升温到220℃后保留时间5min.
2 结果与讨论
2.1 不同工作方式对降解效果的影响
2.1.1 甲苯降解性能 连续降解法与吸附存储-间歇放电法的甲苯去除率分别为 82%和 100% (SED=317J/L,反应温度 30℃).可见,吸附存储-间歇放电法的甲苯去除率明显高于连续降解法.吸附存储-间歇放电过程中,在反应器出口未检测到甲苯,故认为在此条件下,甲苯可被完全氧化转化.在类似间歇放电的研究中,有学者发现开启放电之后会有部分甲苯脱附出来[7,9],在本研究中未观测到甲苯的脱附.这可能是因为本研究甲苯吸附量在催化剂的穿透容量以内,此时催化剂表面与甲苯分子之间的结合力较强[10];同时本实验中等离子体放电的能量密度较高(317J/L),在催化剂装填区域形成了强氧化环境,即使有部分甲苯分子从催化剂表面脱附出来也被快速氧化分解,从而在出口未检测到甲苯.
由图 3可见,相对于连续降解法,吸附存储-间歇放电法的碳平衡以及 CO2选择性大幅度的提高.吸附存储-间歇放电法的碳平衡从 26%大幅提升到了50%,CO2选择性也从62%提升到了82%.连续降解法的甲苯去除率高达80%,但碳平衡仅26%,说明部分甲苯并未完全分解,仅不完全氧化为中间产物堆积在催化剂表面.在吸附存储-间歇放电法中,在吸附存储阶段对甲苯进行预吸附,延长了甲苯在反应器中的停留时间,从而提升了甲苯分子与等离子体放电产生的活性物种(e、O3、O*、OH-等)接触几率.同时,该方法中反应主要是吸附在催化剂表面的甲苯与气相中的活性物种进行反应,能够更加有效地的利用等离子体产生的活性物种.因此,吸附存储-间歇放电法降解甲苯活性较高.

图3 两种降解方式的碳平衡和CO2选择性Fig.3 Carbon balance and CO2selectivity of two degradation method(SED 317J/L, reaction temperature 30℃)
2.1.2 反应后催化剂表面中间产物分析 选取两种降解方式反应后的 SBA-15催化剂,用 CS2溶解GC-MS分析其表面有机中间产物,GC-MS谱图见图4(其中未标示峰为背景杂峰).连续降解法选择反应稳定后的催化剂;吸附存储-间歇放电法是在经过与2.1.1中相同条件的吸附阶段之后,分别选取放电0,5,25,45,65min的催化剂.图4a为保留时间 4~32min的谱图,4b为保留时间7~32min的放大谱图.
仅吸附甲苯未开启放电的催化剂表面没有有机副产物,开启等离子体放电之后,甲苯的降解生成中间产物.连续降解法的催化剂表面检测到了大量的有机中间产物,如苯甲酸、苯甲醛、苯甲醇以及硝基苯酚等,该结果与其他类似研究观测到的中间产物类似[11-12].在吸附存储-间歇放电法中,催化剂表面仅检测到苯甲醛和2-庚烯醇两种中间产物,苯甲醛的数量随着等离子体放电反应的进行逐渐升高,在25min时达到最大值,然后后逐渐降低至低于检测限;2-庚烯醇的数量在放电之后 5min时最高,随着反应的进行逐渐降低至低于检测限.
在连续降解法中,由于降解气体的连续通入,甲苯分子尚未完全降解,又不断有甲苯分子通入,系统无法提供足够的能量将中间产物完全氧化,从而会中间产物在催化剂表面堆积,这将覆盖催化剂表面的活性位点,进一步导致其降解活性变低,这正是前文中连续降解法碳平衡过低的原因.在吸附存储-间歇放电法中,仅是阶段性地降解存储于催化剂中的甲苯,能够更高效地利用等离子体放电产生的活性物种,降解过程中产生的中间产物能够迅速被进一步降解为 COχ,从活性位中脱离出来,从而保持催化剂表面的活性位点暴露[13],故而其催化活性较高.
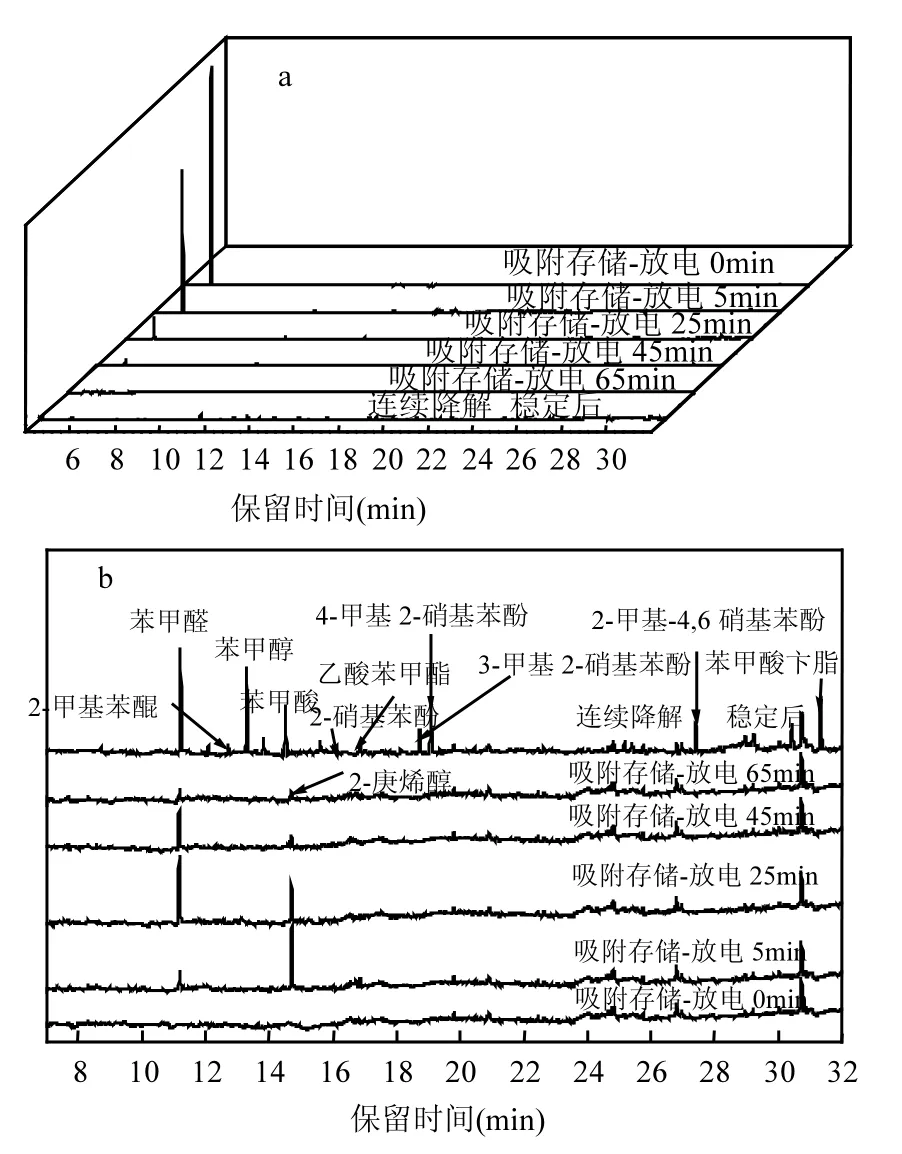
图4 两种工作方式催化剂表面有机中间产物的GC-MS谱图Fig.4 GC-MS spectra of Intermediates on the catalyst surface of two operation modes(SED 317J/L,reaction temperature 30℃,catalyst SBA-15)
2.1.3 等离子体催化降解甲苯的路径分析 等离子体催化降解甲苯主要有3种路径[11],即高能电子解离、电荷转移和活性物种轰击,参与反应的活性物种包括高能电子、H·、OH·、NO2·、O3及其分解产生的O*等.结合本研究中观测到的中间产物(详细物质组分见表1),甲苯在等离子体下的氧化分解路径的主要有 3种(图 5):路径一,苯环上甲基的脱氢,继而在O*和OH·的作用下形成了苯甲醇,苯甲醇再进一步被氧化为苯甲醛和苯甲酸;路径 2,苯环上的C——C键和苯环与甲基之间的C——C键断裂,在O*、OH·和NO2·的作用逐渐形成硝基苯酚类物质.路径 3,苯环开环后逐步氧化为 COχ、H2O等,苯环的开环包括甲苯上的苯环以及前 2种路径中生成芳香族中间产物的开环.
甲苯分子上甲基的C—C键键能为3.7eV,苯环上的C—C键键能为4.3eV、甲基和苯环之间的 C—C键键能为 4.4eV、苯环上的 C—C键键能为5.0~5.3eV、C=C键键能为5.5eV[14],等离子体放电产生的高能粒子的能量范围在1~10eV[15].
运用连续降解法氧化降解甲苯的路径主要是路径 1、2、3.甲苯上甲基的 C—H键能最低(3.7eV),苯环上的甲基脱氢是甲苯氧化的首要路径[15],该方法中催化剂表面有机中间产物中苯甲酸、苯甲醛和苯甲醛占多数(表1),路径1是3种路径中主要路径.由于该方法连续降解的放电方式,甲苯分子并未被完全降解,在催化剂表面检测到了大量的芳香族有机副产物,苯环上的C——C键和C=C键键能最高(5.0~5.3eV,5.5eV),甲苯氧化分解反应主要停止在了苯环开环这一步骤.
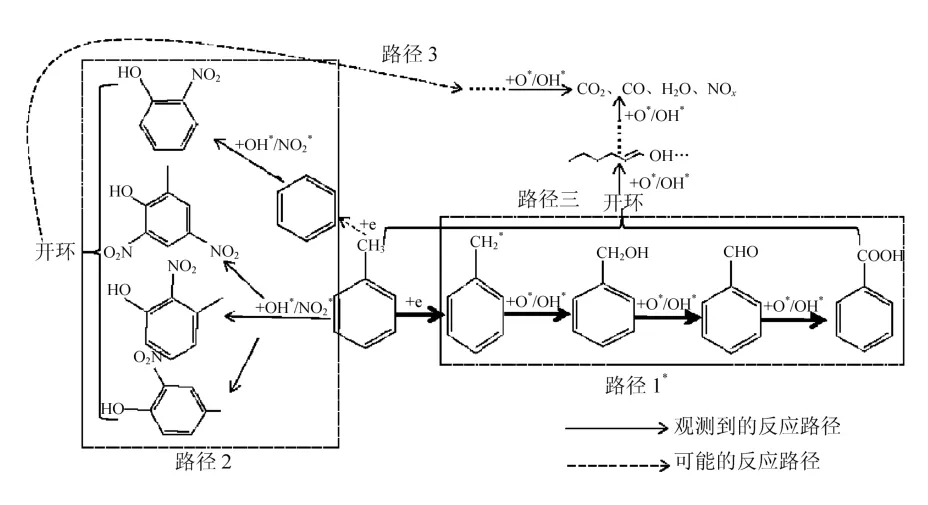
图5 甲苯等离子体催化降解路径假想图(*关键步骤)Fig.5 Pathways of toluene degradation by plasma catalysis(*key steps)
运用吸附存储-间歇放电法氧化降解甲苯路径主要是路径1、3.由于其间歇性降解的放电方式,能够更有效地利用等离子体产生的活性物种,甲苯氧化的中间产物能够迅速氧化分解,故该方法中催化剂表面仅检测到两种物质:苯甲醛和2-庚烯醇.在该方法中,路径2被大大弱化,主要以路径1、3为主.
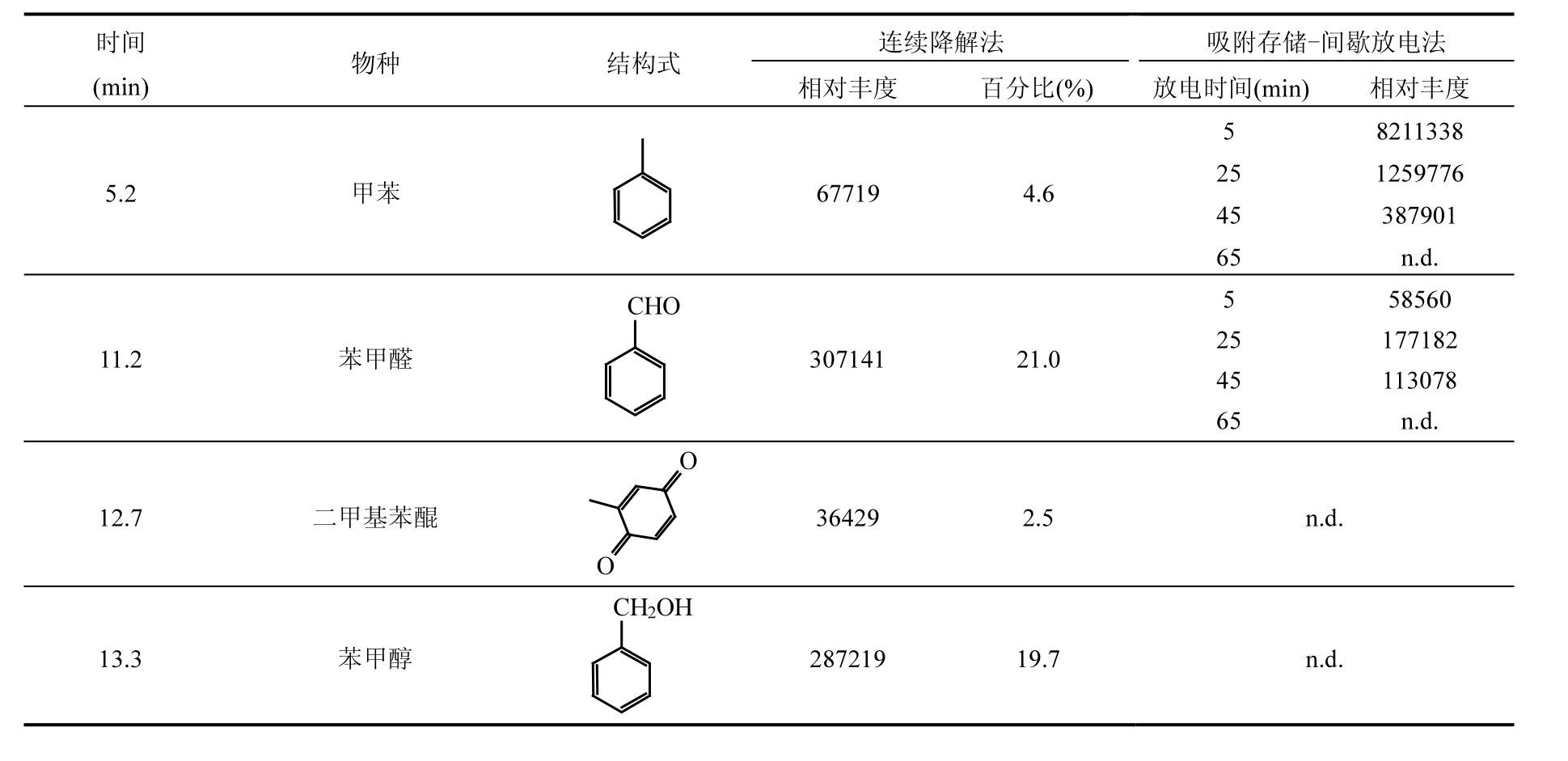
表1 两种工作方式的催化剂表面有机中间产物Table 1 Organic Intermediates on the catalyst surface of two operation modes
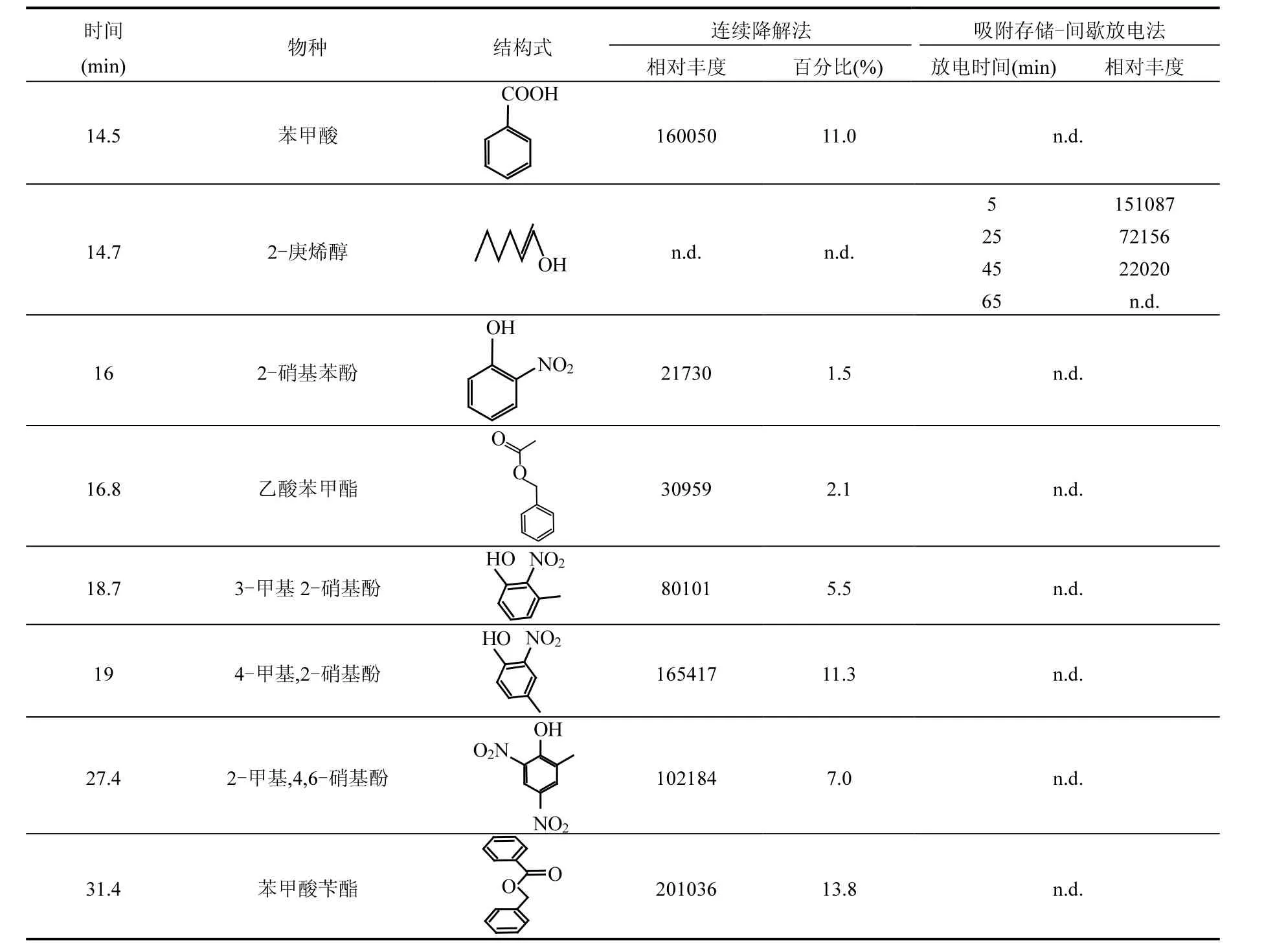
续表1
综上所述,等离子体催化氧化甲苯的控制步骤是:苯甲醛进一步氧化分解,继而打开苯环.其中苯甲醛是关键物种.
2.2 吸附存储-间歇放电法中催化剂对甲苯降解的影响
2.2.1 降解过程中的气相产物的变化 运用吸附存储-间歇放电法,研究SBA-15、Mn/SBA-15、Ag/SBA-15、AgMn/SBA-15 4种催化剂对甲苯降解的影响.其图6为反应过程中,放电反应器出口的甲苯、CO、CO2浓度.在放电销毁阶段,反应器出口未检测到甲苯,说明存储在催化剂当中的甲苯逐渐被完全氧化分解.开启等离子体放电后,CO2和CO均达到急剧升高,在20min左右达到最大值,然后逐渐降低至稳定.65min以后,CO浓度逐渐趋近与零,但CO2浓度仍有30×10-6左右,可能是前期降解甲苯生成的 CO2吸附在催化剂表面,随着甲苯完全分解后,部分CO2逐渐脱附出来[9].
2.2.2 降解过程中催化剂表面有机物种的变化 研究吸附储存-放电销毁法中甲苯的降解过程中催化剂表面有机物种的变化,选去 AgMn/ SBA-15催化剂,在 2.1相同的反应条件下,分别取放电时间0,5,25,45,65min的催化剂,用CS2溶解GC-MS分析表面有机物质,其谱图如图7所示.图7a为保留时间4~32min的谱图,7b为保留时间7~32min的放大谱图.图中保留时间5.2min、11.2min分别为甲苯和苯甲醛.如图,开启等离子体放电之后,0~25min时催化剂表面的甲苯的数量急剧降低,45min时催化剂表面仅有极少量甲苯,在65min时催化剂表面已完全检测不到有机物种.对应2.1中的气相产物,在0~25min时,大量的甲苯被分解,此阶段气相中CO2、CO的浓度急剧上升至最高.25min后,随着大部分甲苯已被氧化分解,CO2、CO的浓度开始迅速降低.65min时,催化剂表面已观测不到有机产物,这也验证了2.1中65min后持续存在的少量的CO2并非未完全降解的中间产物降解生成,而是前阶段吸附在催化剂中的CO2逐渐脱附出来.
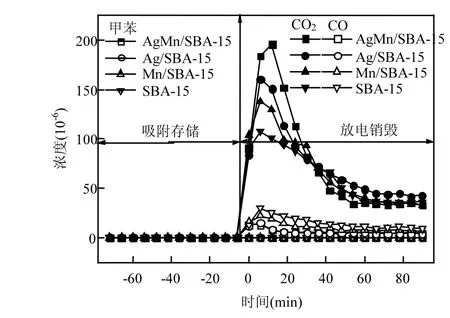
图6 装载四种催化剂反应器出口的甲苯、CO2、CO浓度Fig.6 Concentration of toluene、CO and CO2at outlet of reactor of four catalysts (SED 317J/L, reaction temperature 30℃)
AgMn/SBA-15催化剂降解甲苯全过程中,催化剂表面的副产物仅有苯甲醛,SBA-15催化剂降解过程中生成的2-庚烯醇未检出,可能是由于Ag、Mn负载之后,甲苯氧化的速率加快,苯环开环产物2-庚烯醇能够迅速被氧化分解,因此催化剂表面仅有苯甲醛被检测到,这也进一步验证了苯甲醛是吸附存储-间歇放电法氧化降解甲苯的关键物种.
2.2.3 反应副产物:CO和臭氧 图8为4种催化剂在吸附存储-间歇放电法中 CO2选择性,结合前文中图6,4种催化剂的甲苯氧化速率和CO2选择性顺序均为:AgMn/SBA-15>Ag/SBA-15> Mn/SBA-15>SBA-15.在负载了 Ag、Mn之后,其甲苯氧化速率和 CO2选择性较未负载的SBA-15均明显提升,其中AgMn/SBA-15较未负载的SBA-15的CO2选择性由80%提高到95%,这与负载了 Ag、Mn之后提高了催化剂分解臭氧的性能有关.Ag-Mn-O催化剂具有优良的氧化还原能力、储氧性能和表面氧流动性能等,在一定的条件下,能够产生活性氧参与甲苯的降解,从而促进了反应的进行[15].
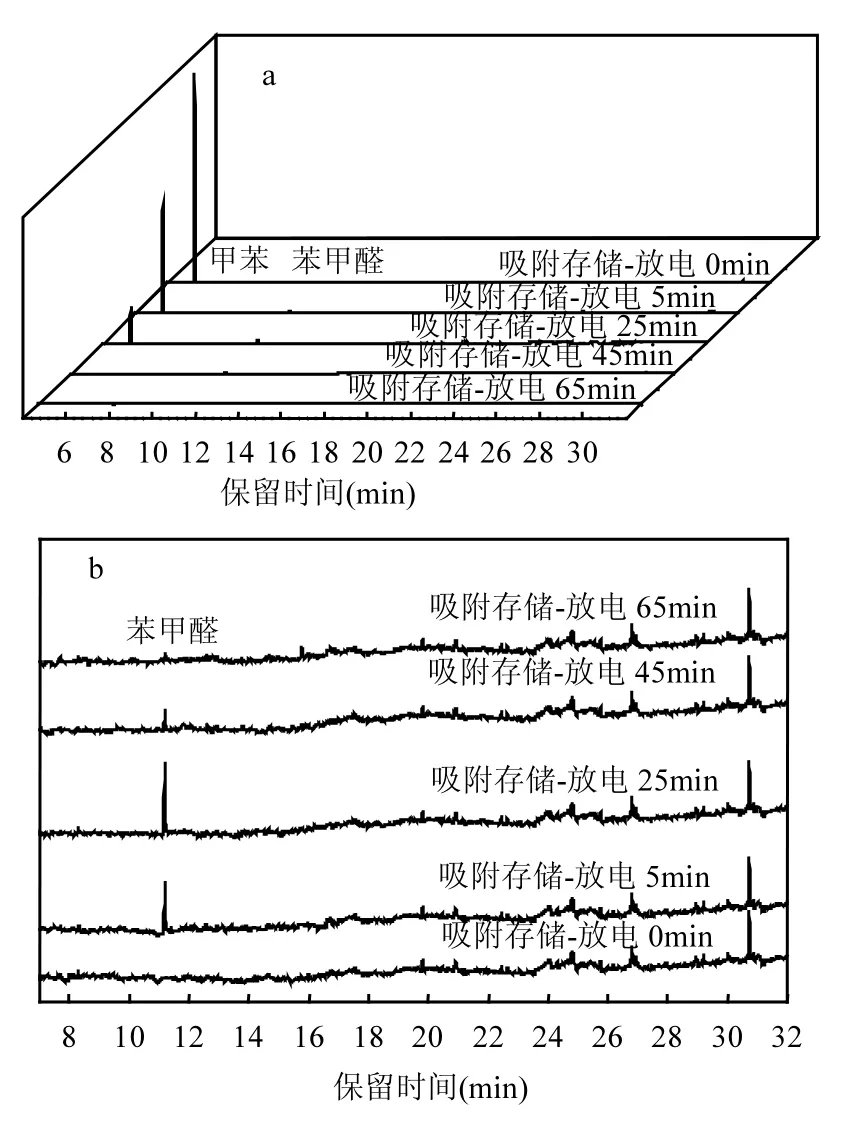
图7 吸附存储-间歇放电法催化剂表面有机物种变化GC-MSFig.7 GC-MS spectra of the change of the intermediates in the process of adsorptive storage- intermittent discharge (SED 317J/L,reaction temperature 30℃, catalyst AgMn /SBA-15)
为了对比催化剂对臭氧的分解能力,采用吸附存储阶段预吸附甲苯与未吸附甲苯两种方式,测定四种催化剂在放电销毁阶段反应器出口的臭氧浓度,结果如图9所示.催化剂预吸附甲苯之后的放电过程中臭氧浓度显著降低,这是由于催化剂分解甲苯消耗了部分臭氧.当未吸附甲苯时,臭氧浓度较高且稳定.当催化剂吸附了甲苯之后,随着吸附存储的甲苯被氧化分解,臭氧浓度逐渐升高,最后稳定到未吸附甲苯时的臭氧浓度. Ag、Mn的负载大大提高了催化剂分解臭氧的能力,AgMn复合催化剂的降解臭氧能力最强,运用该催化剂反应过程中基本检测不到臭氧.四种催化剂分解臭氧的能力顺序为 AgMn/SBA-15> Mn/SBA-15>Ag/SBA-15>SBA-15,其与催化剂氧化甲苯性能的顺序一致.臭氧在催化剂的表面分解为活性氧物种,其公认的一个反应式[17]为:
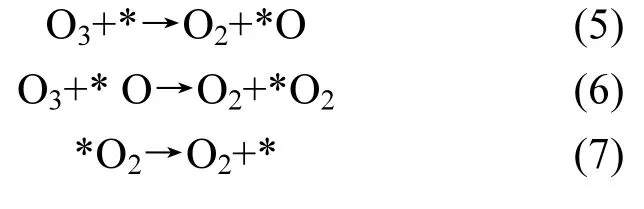
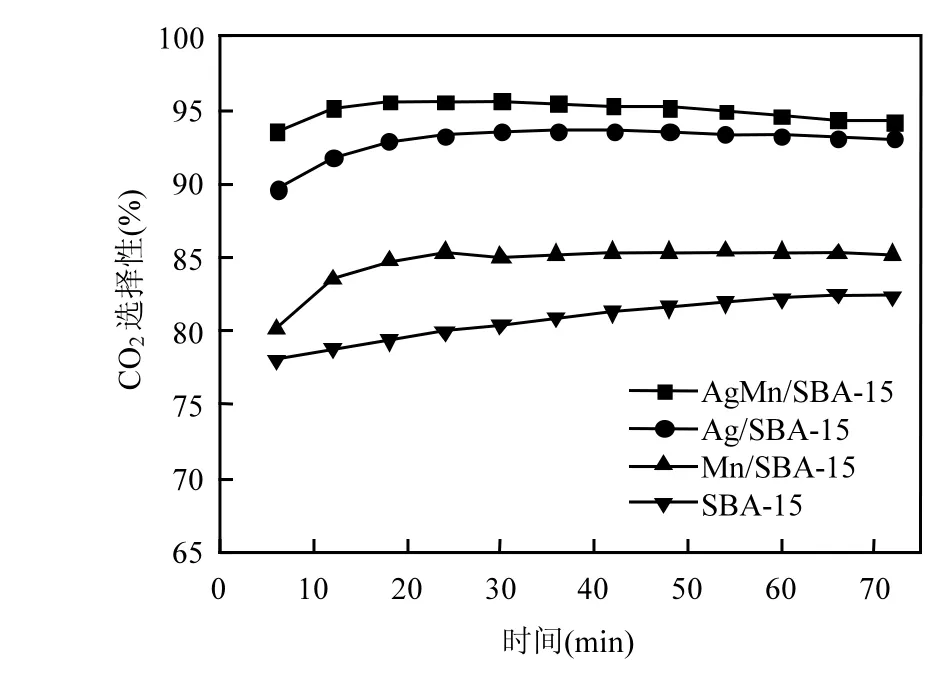
图8 结合不同催化剂的吸附存储—间歇放电甲苯氧化的CO2选择性Fig.8 CO2selectivity of adsorptive storage-intermittent discharge method combined with different catalysis

图9 四种催化剂的臭氧分解性能Fig.9 Ozone decomposition effect of four catalysis (SED 317J/L, reaction temperature 30℃)
式中:*是催化剂表面的活性位,甲苯与活性氧原子的反应速率常数k=7.6×10-14molecule /(cm3·s)远高于臭氧与甲苯的反应速率常数 k=3.9× 10-22molecule/(cm3·s)[18],臭氧被分解地越多,产生的活性原子氧也就越多,就有更多的活性氧参与到甲苯的降解过程中,催化剂表面存储的甲苯在活性原子氧的作用下分解是其降解过程的一个重要步骤,故在放电条件一定的前提下,催化剂分解臭氧的能力与催化降解的甲苯的速率正相关[19].因此,Ag、Mn的负载提高了臭氧分解能力,从而强化了甲苯的氧化效果.
其中Mn/SBA-15催化剂氧化臭氧的能力略强于Ag/SBA-15,但是其氧化甲苯的速度却低于Ag/SBA-15.可能是由于 Ag的存在能够促进氧化中间产物(HCOOH、CO等)的完全氧化为CO2[20],这是在Ag与Mn降解臭氧能力大体相当的情况下,Ag的负载可以进一步把中间产物完全氧化为CO2,故而Ag/SBA-15催化剂CO2选择性高于Mn/SBA-15.
3 结论
3.1 相对传统连续降解法,吸附存储-间歇放电法的甲苯降解率、碳平衡与CO2选择性由82%、26%、62%明显提升至 100%、50%、82%,且催化剂表面的中间产物数量由 10种减少至 2种,其主要原因是在该方法中催化剂能够更加有效地利用等离子体产生的活性物种.
3.2 考察两种降解方式降解甲苯催化剂表面中间产物的变化,苯甲醛进一步氧化分解进而打开苯环是等离子体催化降解甲苯的控制步骤,苯甲醛是其关键物种.
3.3 在吸附存储-间歇放电法净化低浓度甲苯中,Ag、Mn的负载促进了 2-庚烯醇的氧化催化,AgMn/SBA-15拥有最好的碳平衡、CO2选择性.这与Ag、Mn的负载提高了催化剂分解臭氧的性能相关.
[1] Qiu K Q, Yang L X, Lin J M, et al. Historical industrial emissions of non-methane volatile organic compounds in China for the period of 1980-2010 [J]. Atmospheric Environment, 2014,86: 102-112.
[2] 魏长宽,朱天乐,樊 星,等.非热等离子体与催化相结合去除气象低浓度苯系物 [J]. 环境科学学报, 2008,28(4):676-680.
[3] Durme J V, Dewulf J, Leys C, et al. Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: A review [J]. Applied Catalysis B: Environmental, 2008,78:324-333.
[4] Song Y-H, Kim S-J, Choi K-I. Effects of adsorption and temperature on a nonthermal plasma process for removing VOCs [J]. Journal of Electrostatics, 2002,55:189-201.
[5] Zhao D Z, Li X S, Shi C, et al. Low-concentration formaldehyde removal from air using a cycled storage–discharge (CSD) plasma catalytic process [J]. Chemical Engineering Science, 2011,66: 3922-3929.
[6] Zhao D Z, Shi C, Li X S, et al. Enhanced effect of water vapor on complete oxidation of formaldehyde in airwith ozone over MnOx catalysts at room temperature [J]. Journal of Hazardous Materials, 2012,239-240:362-369.
[7] Kuroki T, Fujioka T, Kawabata R, et al. Regeneration of honeycomb zeolite by nonthermal plasma desorption of toluene [J]. IEEE Transation on Industry Appications, 2009,45: 10-15.
[8] Okubo M, Inoue M, Kuroki T, et al. NOxreduction aftertreatment system using nitrogen nonthermal plasma desorption [J]. IEEE Transation on Industry Appications, 2005, 41:891-899.
[9] Mok Y S, Kim D H. Treatment of toluene by using adsorption and nonthermal plasma oxidation process [J]. Current Applied Physics, 2011,11:S58-S62.
[10] Huang Y P, His H C, Liu S C. Preparation of spherical activated phenol-formaldehyde beads from bamboo tar for adsorption of toluene [J]. Journal of the Air and Waste Management Association, 2013,63:8,977-983.
[11] Huang H B, Ye D Q, Dennis Y C, et al. Byproducts and pathways of toluene destruction via plasma-catalysis [J]. Journal of Molecular Catalysis A: Chemical, 2011,87-93.
[12] Durme J V, Dewulf J, Sysmans W, et al. Abatement and degradation pathways of toluene in indoor air by positive corona discharge [J]. Chemosphere, 2007,68:1821-1829.
[13] Sekiguchi k, Sanada A, Sakamoto K. Degradation of toluene with an ozone-decomposition catalyst in the presence of ozone, and the combined effect of TiO2addition [J]. Catalysis Communications, 2003,4:247-252.
[14] Kohno H, Berezin A A, Chang J S, et al. Destruction of volatile organic compounds used in a semiconductor industry by a capillary tube discharge reactor [J]. IEEE Transaction on Industry Applications, 1998,5(34):953-966.
[15] Guo Y F, Ye D Y, Chen K F. Toluene decomposition using a wire-plate dielectric barrier discharge reactor with manganese oxide catalyst in situ [J]. Journal of Molecular Catalysis A:Chemical, 2006,245:93-100.
[16] Ogata A, Ito D, Mizuno K, et al. Effect of coexisting components on aromatic decomposition in a packed-bed plasma reactor [J]. Applied Catalysis A: General, 2002,236:9-15.
[17] Kim H H, Ogata A, Futamura S. Oxygen partial pressuredependent behavior of various catalysts for the total oxidation of VOCs using cycled system of adsorption and oxygen plasma [J]. Applied Catalysis Bdd: Environmental, 2008,79:356-367.
[18] Durme J V, Dewulf J, Sysmans W, et al. Abatement and degradation pathways of toluene in indoor air by positive corona discharge [J]. Chemopsphere, 2007,68:1821-1829.
[19] 吴军良,夏启斌,刘治猛,等.Mn、Fe和Cu氧化物在低温等离子体催化氧化甲苯体系中的活性比较 [J]. 功能材料, 2012,10(43): 1332-1340.
[20] Kim H H, Sugasawa M, Hirata H, et al. Ozone-assisted catalysis of toluene with layere ZSM-5 and Ag/ZSM-5 zeolites [J]. Plasma Chem. Plasma Process, 2013,33:1083-1098.
Reaction process of toluene oxidation by adsorptive storage-intermittent discharge method.
WANG Pei-tao1, HE
Meng-lin1, LU Mei-juan1, HUANG Rong1, FAN Wei-long1, WU Jun-liang1,2, YE Dai-qi1,2,3*(1.College of Environmental and Energy, South China University of Technology, Guangzhou 510006, China;2.Guangdong Provincial Key Laboratory of Pollution Control and Atmospheric Environment, South China University of Technology, Guangzhou 510006, China;3.Air Pollution Control of Guangdong University Engineering Technology Research Center, South China University of Technology, Guangzhou 510006, China). China Environmental Science, 2014,34(12):3047~3055
The effect of continuous discharge and adsorptive storage-intermittent discharge combined with SBA-15on toluene decomposition was investigated under ambient pressure and temperature. The results showed that adsorptive storage-intermittent discharge method exhibited higher toluene conversion, CO2selectivity and better carbon balance compared to the former. The evolution of the intermediate products on the catalyst surfaces with time were also analysed by GC-MS in both methods. The results indicated that formaldehyde oxidation was the rate-determining step for toluene decomposition. Then, the activities of four different catalysts, SBA-15, Mn/SBA-15, Ag/SBA-15and AgMn/SBA-15were tested with dsorptive storage-intermittent discharge mode. AgMn/SBA-15catalyst exhibited the highest CO2selectivity and the best carbon balance among the tested catalysts, which may be due to 2-heptene alcohol oxidation catalyzed by the addition of Ag and Mn.
non-thermal plasma catalysis;toluene;continuously degradation;adsorptive storage-intermittent discharge;intermediate product;degradative pathway
X511
A
1000-6923(2014)12-3047-09
王沛涛(1988-),男,河南三门峡人,华南理工大学硕士研究生,主要从事挥发性有机污染物控制研究.
2014-03-25
国家自然科学基金(51378218,U1201231,50978103),国家“863”计划(2013AA065005)
* 责任作者, 教授, cedqye@scut.edu.cn
猜你喜欢
体育科技文献通报(2022年3期)2022-05-23
煤气与热力(2022年4期)2022-05-23
昆明医科大学学报(2021年6期)2021-07-31
空间科学学报(2021年6期)2021-03-09
中华养生保健(2020年9期)2021-01-18
石油化工技术与经济(2020年4期)2020-09-15
无机化学学报(2019年2期)2019-02-27
北京航空航天大学学报(2017年7期)2017-11-24
北京航空航天大学学报(2017年7期)2017-11-24
北京航空航天大学学报(2017年2期)2017-11-24
