
胰蛋白酶原基因缺失突变导致早发型自身免疫性相关的多器官多发囊肿的研究
2014-04-14 05:41徐志峰林寿榕刘奇才陈瑞庆林丽清翁少煌郜峰庄则豪陈金通
实用检验医师杂志 2014年2期
徐志峰 林寿榕 刘奇才 陈瑞庆 林丽清 翁少煌 郜峰 庄则豪 陈金通
临床研究
胰蛋白酶原基因缺失突变导致早发型自身免疫性相关的多器官多发囊肿的研究
徐志峰 林寿榕 刘奇才 陈瑞庆 林丽清 翁少煌 郜峰 庄则豪 陈金通
目的 探讨由胰蛋白酶原基因(cationic trypsinogen,PRSS1)突变引发的早发型自身免疫性相关的多器官多发囊肿及其致病机制。方法采用DNA全长测序技术分析PRSS1、囊性纤维化跨膜通道调节因子(cystic fibrosis transmembrane conductance regulator,CFTR)、丝氨酸蛋白酶抑制剂Kazal 1型(serine protease inhibitor Kazal type 1,SPINK1)、蛋白激酶D(protein kinase D,PKD)1和PKD2等胰腺炎和多囊性病变相关基因的所有外显子及其侧翼内含子剪切区域,确定DNA和cDNA序列的变异,通过与家系内部和正常对照的比较分析,对检测到的变异是否与疾病相关进行探讨,并构建突变体表达体系进行功能学验证,同时对患者的肺、肝、胰腺等穿刺样本进行免疫组织化学和特殊染色。结果在2例年轻的自身免疫性胰腺炎患者中首次发现PRSS1基因2号外显子缺失突变生成激活肽缺失型的胰蛋白酶原,并具有生物学活性;肝脏、肺穿刺病理均可见不同程度的淋巴细胞和浆细胞浸润,肺组织病理显示弹力纤维、网状纤维明显减少;患者表现为多脏器多囊性病变,血清胰蛋白酶、弹力蛋白酶、AAT显著增高。使用糖皮质激素治疗有效。结论PRSS1:c.1300_1304 del CCCAG是引发早发型自身免疫性胰腺炎的新突变形式,并与多器官囊肿关系密切。
自身免疫性胰腺炎;PRSS1基因;缺失突变;多囊性,多器官
自身免疫性胰腺炎(autoimmune pancreatitis,AIP)是一个常被忽视的“新”疾病。该病通过激素治疗就可获痊愈,但因其多发于老年男性,以梗阻性黄疸为主要临床表现,因此通常被误诊为胰腺癌或胆管癌,而实施了高风险的手术,这不能不说是一个悲剧!虽然,随着国际上多个诊断标准的确立,AIP的误诊率逐渐降低,但由于对这种“新”疾病发病机制的了解甚少,误诊现象依然存在。
AIP与普通的胰腺炎不同,病因不是酒精损害、胰腺结石等,而是自身免疫功能异常。血清IgG4增高是Ⅰ型AIP的显著特征。近年来,其筛查和鉴别AIP的价值逐渐在医学界得到认可和关注,武建国等[1]较早在国内系统评价IgG4和自身抗体在鉴别AIP中的作用,另一些学者[2-4]更具体的说明了IgG4的诊断价值和胰蛋白酶原等自身抗体在AIP中发挥的重要作用及其具有的潜在诊断应用价值。
然而,直接导致AIP自身免疫失调的因素仍然未知。已知胰蛋白酶可以作为淋巴细胞的刺激源在机体免疫监视中起重要的作用,胰蛋白酶原基因突变可导致胰蛋白酶原的异位激活,造成淋巴等慢性炎症细胞的浸润,压迫、阻塞腺体造成分泌腺的功能缺陷。这个结果可以解释部分AIP的发生机制,同时也印证了AIP患者中存在胰蛋白酶原和抗胰蛋白酶原抗体的异常表达。本文研究描述的AIP患者其共有的特性是严重的肺部损害,也许是AIP的另一个特征,也是胰蛋白酶原异常激活造成弹力蛋白消耗和淋巴细胞活化的结果,也可能是目前还没有认识到的由酶基因突变之后造成的一种新的自身免疫综合征。
1 资料与方法
1.1 临床资料病例1:患者,男,23岁,因“皮肤巩膜黄染,气促20余天”第4次入院,无发热、腹痛。实验室检查AMY 571 U/L(30~110 U/L),TBIL 413.5 μmol/L(0~19 μmol/L),DBIL 315.5 μmol/L(0~7 μmol/L),ALT 257 U/L(5~40 U/L),AST 304 U/L(5~50 U/L),TBA 283.4 μmol/L(0~10 μmol/L)。经对症处理后,呼吸有所改善,但黄疸不退。CT示胰腺体积增大、肝门区可疑病变、肝内胆管扩张、肺气肿、双肺多发肺大泡。4年前体检发现肝功能异常,无不适,3月前全身皮肤黄染、眼黄、尿黄,食欲稍差,近3年来体重下降约20 kg,无酗酒史。疑诊为α-抗胰蛋白酶(α-antitrypsin,AAT)缺乏症,但随后发现血清AAT反而是正常上限的2倍。血清蛋白电泳:α1-球蛋白5.8%(2.9%~4.9%),β2-球蛋白18.0%(3.2%~6.5%),最后AAT基因测序也排除了AAT缺乏症的诊断。做进一步检查:肝炎标志物阴性,抗核抗体均质型弱阳性,余阴性。
病例2:患者,男,21岁,因“纳差伴上腹痛2 w”入院。半年前出现食欲减退伴有阵发性的上腹隐痛,按胃病治疗无效,2月前出现皮肤巩膜黄染及皮肤瘙痒,尿黄、尿糖阳性,间歇性气喘。CT检查发现胰头肿大、肝外胆管扩张,诊断为“胰腺占位、梗阻性黄疸、肺大泡、糖尿病”。发病期间自觉乏力、1个月体重下降4 kg,既往无饮酒史,其父10年前行肺大泡切除术(有黄疸和气喘,但具体不详),其姑亦有黄疸、胆囊炎、胰腺炎史(TBIL最高113.2 μmol/L)。实验室检查:TBIL 165.3 μmol/L,DBIL 59.6 μmol/L,ALT 98 U/L,AST 41 U/L,总胆汁酸45.2 μmol/L,r-GT 452 U/L,ALP 265 U/L,血糖13.22 mmol/L,肿瘤标志物正常,肝炎标志物阴性。
随机选择520例无血缘关系的健康志愿者作为正常对照组,其中男336例,女184例,年龄47~68岁。
1.2 研究方法
1.2.1 基因组突变分析在患者及其家属和正常受试者知情同意的情况下,使用Tiangen Genomic DNA提取试剂盒(北京,中国)提取患者全血基因组DNA。扩增患者及家系成员和对照者的胰腺炎和多囊性病变相关的PRSS1、CFTR、SPINK1、PKD1和PKD2等基因的所有外显子及其侧翼内含子剪切区域。接着对PCR产物进行纯化、测序,将测序结果与NCBI上公布的参考序列进行比对。
1.2.2 突变体功能学实验将完整的突变型和野生型PRSS1 cDNA序列导入质粒并连接感受态菌:以含胰蛋白酶原基因的重组质粒pQE-Tg2为模板,设计引物进行PCR扩增,正向引物为5′-TGCAATTGTATGGCACCATTCGACGATGATGACAAGAT-3′,反向引物为5′-GAGTCGACTCAGCTAATTAAGCTTAGTG-3′。正向引物和反向引物的两端分别引入Mun I和Sal I酶切位点,挑选单克隆,测序验证,建立表达体系;在胰蛋白酶标准品(R&D公司,美国)监测的情况下,对胰蛋白酶进行分离、纯化和复性。以benzoyl L-arginine ethyl ester为底物,用紫外吸收法测定30 min内D(253 nm)值的变化,酶比活力测定公式为:
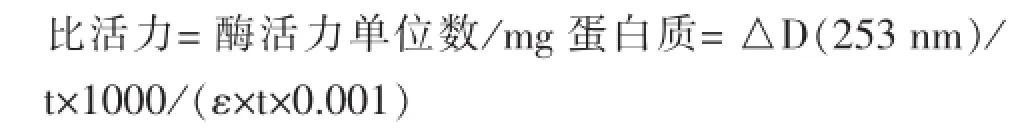
t为时间间隔(min),ε为测定时用的蛋白酶量(μg)。
1.2.3 胰蛋白酶、AAT和弹力蛋白酶检测患者在清晨安静状态留取血清标本连同对照组的样本收集后-80℃冻存。血清弹力蛋白酶、胰蛋白酶原激活肽(trypsinogen activation peptides,TAP)和胰蛋白酶应用ELISA试剂盒(R&D公司,美国)。AAT检测应用BNⅡ特定蛋白分析仪及其配套试剂(柏林,德国)。
1.2.4 穿刺标本免疫组织化学和电镜检测穿刺标本(肝脏、肺)进行特殊染色检测网状纤维等,并进行免疫组织化学(CK、CD3、CD20、CD38、CD68和Vimentin等)分析,HE染色观察淋巴细胞和浆细胞浸润情况。
2 结果
2.1 基因测序分析在病例1和病例2中发现PRSS1基因新的突变形式(c.1300_1304 del CCCAG)(图1),这种移码突变导致PRSS1基因2号外显子大片段缺失,缺失片段(14~54aa)包括胰蛋白酶原激活肽(18~23aa)在内的41个氨基酸(图1),产生无胰蛋白酶原激活肽的截短蛋白,这种突变形式不存在于正常对照和家系其他正常成员中。
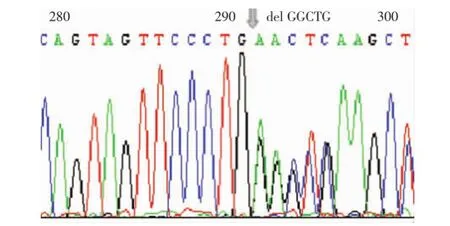
图1 PRSS1基因缺失突变DNA和cDNA测序图
2.2 病理学分析胰腺组织呈典型的自身免疫性胰腺炎特征:弥漫性纤维化、慢性炎性细胞浸润、脉管炎和神经周围炎性细胞浸润(图2A)。浸润浆细胞为IgG4抗体阳性的浆细胞(10~45个/高倍视野)(图2B),电镜扫描显示病变区的淋巴浆细胞浸润和纤维化(图2C)。肺组织病理:符合肺大泡改变,灶区组织细胞结节状增生。免疫组织化学染色结果:CD3T细胞(+),CD20B细胞(+),CD68组织细胞(+),CK上皮(+),Vimentin(+)(图2D)。骨髓病理活检:(骨髓涂片)骨髓增生大致正常,粒系以成熟形态为主,粒系红系比值约为2:1,红系数量形态未见异常,巨核细胞数量为2~4个/高倍视野。免疫组织化学染色结果:CD15、MPO粒系(+)、CD235红系(+)、F8巨核系(+)(图2E)、肝脏病理:镜下见重度慢性大胆管阻塞性淤胆伴胆汁溢出,部分间质纤维组织增生伴慢性炎症浸润及局灶性胆汁梗死。肝脏组织电镜:肝细胞内见大量胆色素颗粒,肝细胞间毛细胆管扩张、瘀胆,部分肝细胞间胶原纤维增生,肝kupffer细胞增生明显,细胞内大量胆色素颗粒及部分髓样结构,肝细胞内及窦周间隙见少量淋巴细胞浸润。汇管区扩大,未见小胆管,胶原纤维增生,并见少量炎症细胞浸润。考虑胆汁淤积性肝损害,由于肝kupffer细胞内可见较多髓样结构,故考虑Niemann-Pick病可能性大。肺组织中肺泡间隔的弹力蛋白严重消耗,弹力纤维断裂(图2F)。
2.3 影像学资料肝脏MRI:不均匀脂肪肝;肝内多发长T1长T2信号灶;肝胃间隙内结节影,肿大淋巴结。胸部CT平扫:双侧气胸引流术后,胸壁皮下气肿;左下肺炎并肺不张;双侧多发肺大泡;肝内多发低密度灶;肺CT平扫:肺气肿;双肺多发肺大泡;左肺下叶小结节;肝门区可疑病变,肝内胆管扩张(图3)。
2.4 血清酶学和相关临床指标患者血清胰蛋白酶、AAT、弹力蛋白酶增高,发病期间体重下降明显,纤维化指标:透明质酸、Ⅳ型胶原、层黏连蛋白和Ⅲ型前胶原N端肽均显著性增高。实验室检查示:2例患者IgG4 16.396 g/L(波动范围2.215~22.352 g/L)和IgG4 8.153g/L(波动范围3.102~15.041 g/L)。泼尼松龙治疗6个月均好转。
2.5 突变体表达产物活性测定紫外分光光度计测量△D(253 nm)来检测肠激酶活化前后的胰蛋白酶原活性。连续观察30 min,发现突变体表达产物经肠激酶活化前后的活性差异无统计学意义,而且D(253 nm)每增加0.002所用的△t为160-180 s,根据公式计算复性重组胰蛋白酶原肠激酶活化前后的胰蛋白酶比活力为65~75 BAEE U/mg;野生型胰蛋白酶原经肠激酶活化后比活力为123~165 BAEE U/mg,以该方法测得猪天然胰蛋白酶标准品的比活力为1080 BAEE U/mg。这说明了突变体的活性降低,机体可能通过增加合成代偿机制来弥补活性的缺陷,但是增多的突变型的胰蛋白酶原本身不稳定,在胰腺内部可以通过自激活途径活化,从而造成胰蛋白酶原的异位激活引起胰腺炎。同时由于肺等器官也表达有胰蛋白酶原,因此也造成相应器官的炎症和囊性病变。
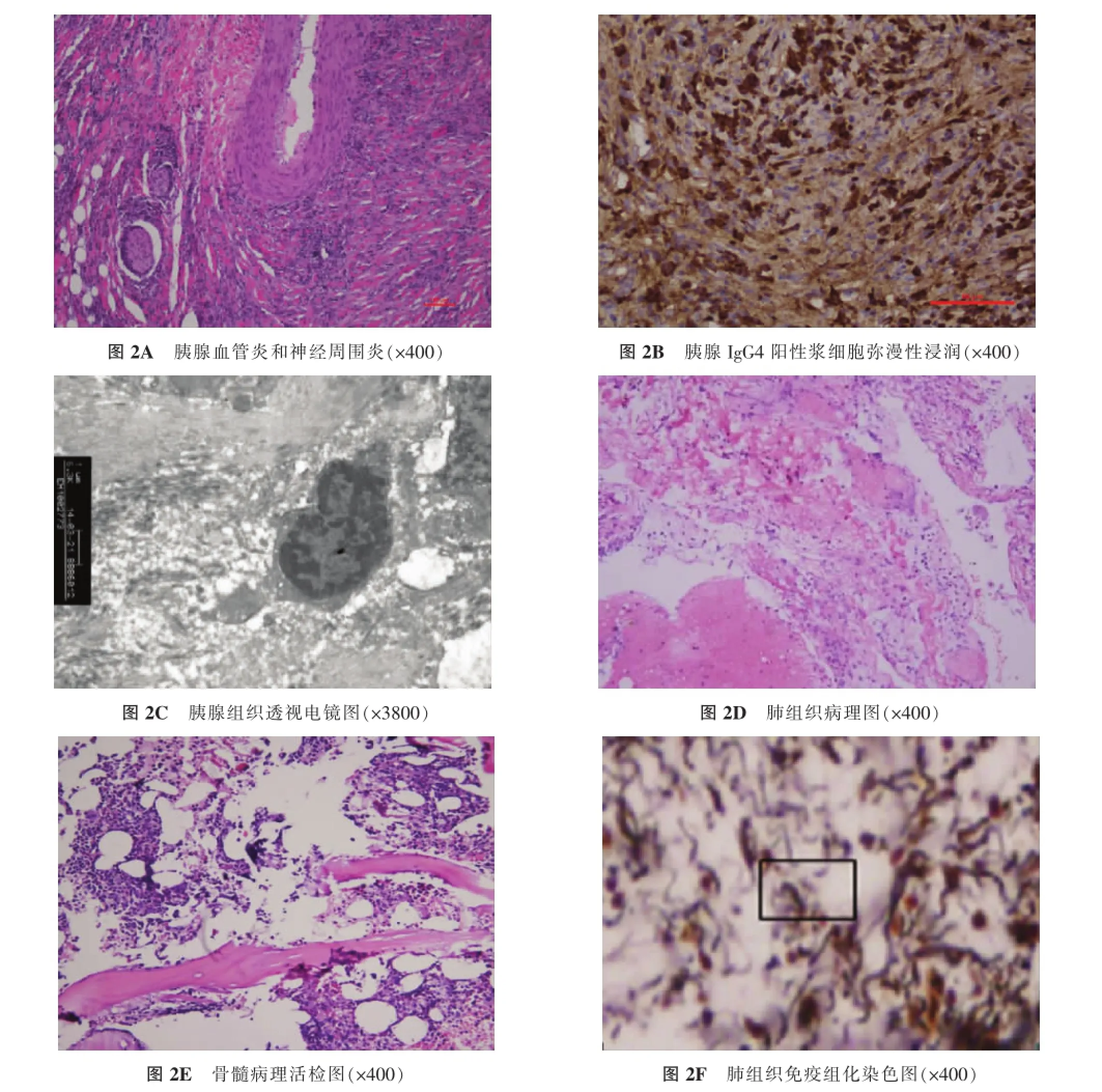
图2 肝肺组织病理学检查和特殊染色、免疫组化结果
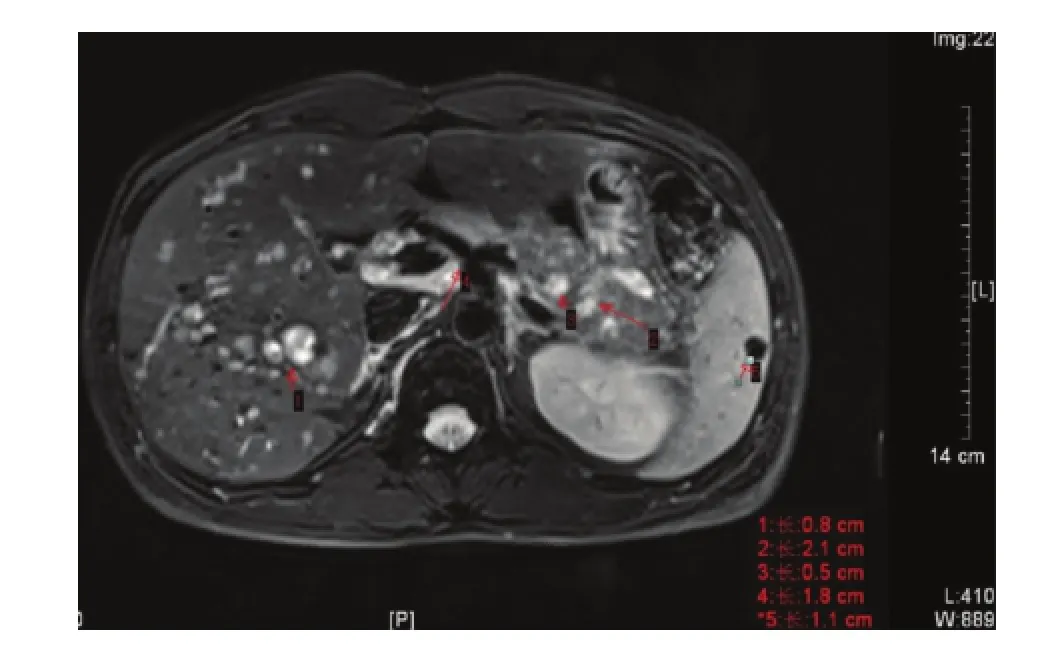
图3 肝胆胰脾多发囊肿、腹膜后淋巴结肿大和肺大泡影像图
3 讨论
已经明确的基因突变引发多器官损伤中,以囊性纤维化(cystic fibrosis,CF)研究居多,包括肺纤维化、胰腺炎和30%的病例合并肝损伤[5-7],然而CF在汉族人群中相对罕见。本文研究从基因组、cDNA、突变体功能和患者的临床资料等方面阐述由胰蛋白酶原基因缺失突变而造成胰蛋白酶原提前激活,从而引发AIP相关的肺肝胆胰脾多器官多发囊肿合并黄疸的病理过程。
AIP发病机制至今不明,有研究显示遗传因素在AIP发病机制中发挥着主导作用。日本人群中AIP患者单倍体基因DRBI*0405-DQBl*0401显著高于其他慢性胰腺炎患者[8-11]。Parkdo等[12]发现组织相容性白细胞抗原单倍体DQβ1第57位天冬氨酸置换与AIP的复发显著相关。而胰蛋白酶原基因嵌插在TCR β位点中(也就是胰蛋白酶原基因与TCR β基因共定位)这种现象不管在人、小鼠还是鸡中也是保守的,这可能预示着胰蛋白酶原和TCR β拥有共同的免疫功能或调节因素,正如主要组织相容性复合物的Ⅰ和Ⅱ类基因,他们拥有相似的组织关系一样[13-15]。
PRSS1基因突变起初是在遗传性胰腺炎中发现的,已经报道[16-17]的胰蛋白酶原基因变异形式有200多种,与胰腺炎相关的突变主要集中在2号外显子和3号外显子。正常情况下,胰蛋白酶原在肠激酶作用下从N端切除16~23位的8个氨基酸残基即胰蛋白酶原活化肽,成为具有活性的胰蛋白酶,活性中心由His40、Asp84、Trp193和Ser177构成一个对底物严格限制的“深口袋”,酶底物蛋白带正电荷的Lys和Arg残基刚好与带负电荷的trypsin催化部位底物Asp-189结合。而当TAP因发生移码突变而缺失后,胰蛋白酶原前体从内质网切除signal cleavage site后即成为有活性的胰蛋白酶,从而造成胰蛋白酶原的异位激活而后激活溶酶体酶、弹力蛋白酶等,或者突变的胰蛋白酶可能还通过直接降解弹力蛋白等机制造成腺体破坏、融合。
胰蛋白酶在胰腺、肺、肝、肠道等均有表达,当胰蛋白酶原基因发生缺失突变后,其表达产物失去了TAP的保护而直接具有活性,激活腺体内的溶酶体酶从而引起胰蛋白酶表达的器官损伤。胰蛋白酶原基因与人类的TCR β基因共定位,不排除基因突变引发的免疫反应和器官损伤引起WBC增高,另外患者不断增高的胆汁酸也进一步刺激了WBC增高,AAT不足以中和中性粒细胞弹力蛋白酶,弹力蛋白消耗,腺体破坏、塌陷,器官损伤(图4)。
PRSS1基因突变生成异常的胰蛋白酶原或者胰蛋白酶表达量异常影响其在胰腺中的定位及其分泌过程,可导致胰腺、肝、肺等存在胰蛋白酶表达的组织胰蛋白酶和抗胰蛋白酶活性平衡的失调,失去了对淋巴等炎症细胞的抑制和降低了扩血管效应,而且炎症细胞本身具有嗜神经性,在这些因素综合作用下,微小导管和动静脉阻塞,腺管和胰腺实质内压力升高,胰液流出障碍,消化酶的分泌、活化异常,诱发慢性胰腺炎。
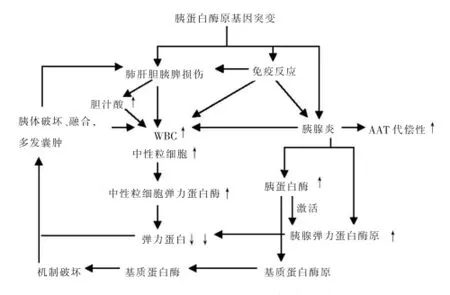
图4 PRSS1缺失突变引发多器官囊性病变的机制图
胰蛋白酶原基因突变及其表达量异常与AIP关系的阐明为揭示内分泌系统与免疫系统之间的相互作用积累了科学依据。但是由于本文研究中的AIP频发的肺大泡和多器官囊肿是其明显的胰腺外的器官受损,也许这是因酶基因突变或活化异常后引发的一种新的自身免疫相关的临床综合征,有待进一步研究。
郑重声明本文的测序图和临床资料已经发表在Current Molecular Medicine,2014,14:340-348上,在其基础上本文继续深入讨论自身免疫性胰腺炎的发病机制和实验室特征以期引起临床工作者的重视。
1 武建国.关注自身免疫性肝病和自身免疫性胰腺炎的实验诊断.临床检验杂志,2007,25:323-325.
2 吴康,沈茜.IgG4检测在自身免疫性胰腺炎诊治中的应用进展.检验医学,2011,26:135-137.
3 孙林,吴炯,郭玮,等.IgG4检测在自身免疫性胰腺炎和IgG4相关性胆管炎诊治中的应用进展.临床检验杂志,2012,30:525-527.
4 宋淑然,刘晓雷,赵建宏.IgG4和自身抗体在自身免疫性胰腺炎诊断中的研究进展.临床检验杂志,2013,31:288-290.
5 Kamisawa T,Egawa N,Nakajima H,et al.Clinical difficulties in the differentiation of autoimmune pancreatitis and pancreatic carcinoma.Am J Gastroenterol,2003,98:2694-2699.
6 Wakabayashi T,Kawaura Y,Satomura Y,et al.Clinical and imaging features of autoimmune pancreatitis with focal pancreatic swelling or mass formation:comparison with so-called tumor-forming pancreatitis and pancreatic carcinoma.Am J Gastroenterol,2003,98:2679-2687.
7 Raina A,Krasinskas AM,Greer JB,et al.Serum immunoglobulin G fraction 4 levels in pancreatic cancer:elevations not associated withautoimmune pancreatitis.Arch Pathol Lab Med,2008,132:48-53.
8 Ghazale A,Chari ST,Smyrk TC,et al.Value of serum IgG4 in the diagnosis of autoimmune pancreatitis and in distinguishing it from pancreatic cancer.Am J Gastroenterol,2007,102:1646-1653.
9 Shimosegawa T,Chari ST,Frulloni L,et al.International consensus diagnostic criteria for autoimmune pancreatitis:guidelines of the International Association of Pancreatology.Pancreas,2011,40:352-358.
10 Creighton J,Lyall R,Wilson DI,et al.Mutations of the cationic trypsinogen gene in patients with chronic pancreatitis.Lancet,1999,354:42-43.
11 Gao F,Li Y,Wang C,et al.Identification of a novel frame-shift mutation in PRSS1 gene in Han patients with autoimmune pancreatitis.Curr Mol Med,2014,14:340-348.
12 Parkdo H,Kim MH,Oh HB,et al.Substitution of aspartic acid at position 57 of the DQ betal affects relapse of autoimmune pancreatitis. Gastroenterology,2008,134:440-446.
13 Kamisawa T,Shimosegawa T,Okazaki K,et al.Standard steroid treatment for autoimmune pancreatitis.Gut,2009,58:1504-1507.
14 Okazaki K,Uchida K,Koyabu M,et al.Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease.J Gastroenterol,2011,46:277-288.
15 Kamisawa T,Shimosegawa T.Pancreas:Histological diagnostic criteria for autoimmune pancreatitis.Nat Rev Gastroenterol Hepatol,2011,9:8-10.
16 Masanori K,Kazushige U,Hideaki M,et al.Analysis of regulatory T cell and IgG4-positive plasma cells among patients of IgG4-related sclerosing cholangitis and autoimmune liver diseases.J Gastroenterol,2010,45:732-741.
17 Durno C,Corey M,Zielenski J,et al.Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis.Gastroenterology,2002,123:1857-1864.
Trypsinogen gene deletion mutation causes early onset autoimmune related pulmonary bulla,hepatic multiple cysts
XU Zhi-feng1,LIN Shou-rong2,LIU Qi-cai2,et al.1Department of Surgery,the First Affiliated Hospital of Fuzhou General Hospital of Nanjing Military,Putian 351100,China2Department of Clinical Laboratory,the First Affiliated Hospital,Fujian Medical University,Fuzhou 350004,China
ObjectiveTo identification of cationic trypsinogen(PRSS1)gene deletion mutation in autoimmune related multiple cysts and its pathogenic mechanism.MethodsAll exons and flanking intron shear region of pancreatitis and polycystic lesions related genes including PRSS1,cystic fibrosis transmembrane conductance regulator(CFTR),serine protease inhibitor Kazal type 1(SPINK1),protein kinase D1(PKD1)and PKD2 were analyzed by DNA sequencing technology.The sequential variation of DNA and cDNA were detected.Whether the variation associated with disease were detected by comparing with family inside and healthy controls.The mutant expression system was constructed and its functional verification was done.At the same time,immunohistochemical and special staining in patients with lung and liver pancreas biopsy samples were executed.ResultsIn two patients with autoimmune pancreatitis,deletion mutant in exon 2 of PRSS1 gene were first found,and it generating activation peptide deletion trypsinogen with biological activity.The liver,lung was lymphocytic and plasma cell infiltration,elastic fibers and reticular fibers decreased the formation of multiple organ polycystic disease.Serum trypsin,elastase and alpha antitrypsin increased significantly.Use of glucocorticoid treatment was effective.ConclusionPRSS1:c.1300_1304 del CCCAG is a new mutationcauses early onset of autoimmune pancreatitis and it correlated with multiple organ cyst closely.
Autoimmune pancreatitis;PRSS1 gene;Mutation;Polycystic,multiple organ
10.3969/j.issn.1674-7151.2014.02.002
2014-01-22)
(本文编辑:杨军)
国家高新技术基础研究863计划(2012AA022604);福建省自然科学基金(2013J1023);福建省医学创新课题(2013-CXB-21);福建省高校杰出青年基金(JA12133)
351100 莆田市,南京军区福州总医院附属第一医院普通外科(徐志峰)
350004 福州市,福建医科大学附属第一医院检验科(林寿榕 刘奇才)
350004 福州市,福建医科大学附属第一医院肝病中心(陈瑞庆)
350004 福州市,福建医科大学药学院(林丽清 翁少煌)
350004 福州市,福建医科大学附属第一医院病理科(郜峰)
350004 福州市,福建医科大学附属第一医院消化科(庄则豪 陈金通)
刘奇才,E-mail:lqc673673673@163.com
猜你喜欢
流行色(2021年8期)2021-11-09
中国民间疗法(2021年13期)2021-08-30
中老年保健(2021年3期)2021-08-22
天津医科大学学报(2021年2期)2021-03-29
现代临床医学(2019年6期)2019-12-07
汉语世界(The World of Chinese)(2019年5期)2019-11-11
科学中国人(2017年36期)2017-06-09
西南医科大学学报(2015年1期)2015-08-22
西南军医(2014年1期)2014-02-03
儿童故事画报(2013年3期)2013-06-24
