
基于磁珠富集法的绣线菊SSR分子标记分离与筛选
2014-03-25 06:32:14张金华GulzarKhan付鹏程雷淑云陈世龙张发起
生物学杂志 2014年3期
张金华,Gulzar Khan,付鹏程,雷淑云,陈世龙,张发起
(1. 中国科学院 高原生物适应与进化重点实验室,中国科学院 西北高原生物研究所,西宁 810001; 2. 青海省作物分子育种重点实验室,西宁 810001;3. 中国科学院大学,北京 100039)
微卫星是DNA短的串联简单重复序列(simple sequence repeat, SSR),一般是由1~6个碱基组成的串联重复DNA序列[1, 2]。研究表明SSR存在于绝大多数的真核生物基因组中[3]。其中最常见的SSR序列为(AC)n,约有5×104~7×105随机分散于整个基因组中,其重复次数和重复程度在不同的物种间高度变化[4]。由于SSR具有高度多态性和共显性等特点,已经广泛应用于遗传图谱构建、基因定位、遗传多样性研究、物种进化与亲缘关系等[5]。
传统的微卫星开发方法是利用菌落原位杂交技术筛选出重组克隆,然后经测序筛选含微卫星序列的DNA片段,构建基因组DNA文库,但该方法耗时费力及筛选效率较低[6]。随着微卫星分离技术的发展,磁珠富集法于1992首次提出,其主要是将酶切产物经PCR扩增后与生物素标记的探针杂交,然后用磁珠富集,从而获得富含SSR序列的片段,从而构建微卫星文库[7]。由于这一方法操作简单且获得SSR分子标记效率高而快速发展。但目前利用磁珠富集法开展绣线菊SSR分子标记的工作还很少见[8]。

图1 菌落PCR检测的琼脂糖电泳图谱(25泳道为分子量标记:DNA maker DL2000)Fig 1 Profile of the PCR amplification of screening of colony (Lane 25 is the DNA molecular maker: DL2000 )
绣线菊亚科(Spiraeoideae)是蔷薇科(Rosaceae)中最古老的亚科,共有22属260余种,中国有8属100余种,全为落叶性(本亚科包括常绿和落叶两大类群,后者为进化类群),绣线菊属(SpriaeaL.)又是绣线菊亚科中最原始的属,在系统进化过程中,衍生出形态各异而亲缘关系紧密的绣线菊种类[9]。高山绣线菊(Spiraeamongolica)和蒙古绣线菊(Spiraeamongolica)同为蔷薇科绣线菊属落叶灌木,多见于海拔1500~4700 m的山坡灌丛等处,是青藏高原地区高山灌丛的主要建群种,对维持高寒草甸生态系统的稳定具有重要作用[10]。在之前对高山绣线菊的谱系地理学研究发现,高山绣线菊在青藏高原台面、东南边缘横断山区等地存在多个冰期避难所,并且没有表现出大规模的种群集体扩张和迁移的现象[11, 12]。但这一结果仅利用叶绿体DNA数据进行分析,由于缺少SSR分子标记而没有进行该方面的检验,不能够完整地展现高山绣线菊的冰期进化历史等问题。因此,本研究利用微卫星富集法从高山绣线菊基因组DNA中分离和获得SSR分子标记,并验证其在蒙古绣线菊的通用性,以期为绣线菊的分类、谱系地理、系统发育等研究提供丰富的SSR分子标记。
1 材料与方法
1.1 材料
研究材料主要采自青海囊谦和四川红原。其中,每个居群随机采集20~27个个体,每个居群内采样个体之间至少间隔100 m,共采集到两种绣线菊4个居群92个个体(表1)。采集植物生长良好的新鲜、幼嫩叶片后,迅速用硅胶干燥并带回实验室于-20℃保存备用。凭证标本存于中国科学院西北高原生物研究所青藏高原生物标本馆(HNWP)。
1.2 高山绣线菊基因组DNA提取
采用改良的CTAB法提取高山绣线菊和蒙古绣线菊基因组DNA,并用乙醇沉淀法对DNA初提母液进行纯化[13]。用紫外/可见光分光光度计NanoDropTM2000c(Thermo Scientific,美国)测定纯化后的基因组DNA浓度,调整DNA总的质量浓度为100 ng/μL,用1%琼脂糖凝胶电泳检测DNA提取结果。
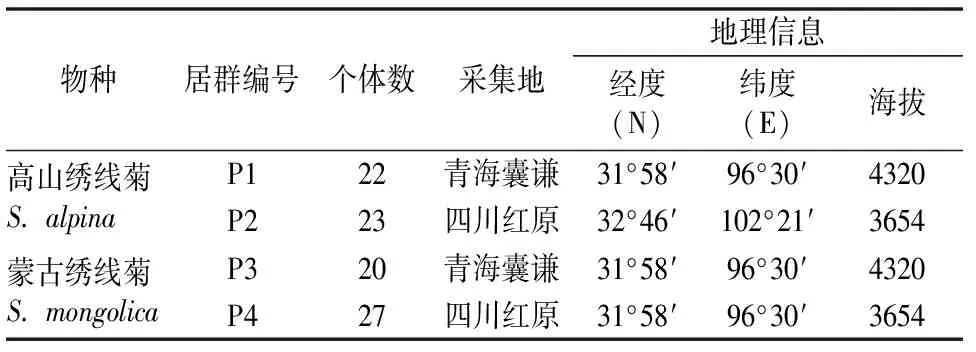
表 1 高山绣线菊和蒙古绣线菊采集信息
1.3 微卫星富集文库的构建
1.3.1 基因组DNA酶切、接头连接及PCR扩增
用MseI内切酶(New England Biolabs,美国)对高山绣线菊的基因组DNA(约200 ng)酶切3~4 h。用1%的琼脂糖凝胶检测酶切结果。将合成的链A(5′-TAC TCA GGA CTC AT-3′)和链B(5′-GAC GAT GAG TCC TGA G-3′)配成20 pmol/μL溶液。分别取链A和B溶液各50 μL于1.5 mL离心管中混合。升温至80℃后,变性5~10 min。在室温下自然冷却1 h,加dd H2O配成终浓度为10 pmol/μL的接头溶液,于-20℃保存。用T4 DNA连接酶(New England Biolabs,美国)将酶切产物与接头溶液于16℃恒温下连接5 h。以此连接产物为模板用简并引物MseⅠ-N [5′-GAT GAG TCC TGA GTA A (N)-3′] 进行PCR扩增,检测接头连接结果。PCR反应采用25 μL体系:连接产物2 μL,2.5 μL的10×PCR Buffer(含1.5 mmol/L MgCl2),0.3 μL 10 mmol/L dNTP,正反引物各1.0 μL(5 pmol/L),1 U Taq DNA聚合酶(TaKara,大连),用ddH2O补足25 μL。PCR扩增反应程序:94℃预变性4 min;30个循环的94℃加热变性1 min,53℃退火45 s,72℃延伸45 s;最后72℃延伸10 min。PCR产物在1.5%的琼脂糖凝胶上进行电泳检测。
1.3.2 探针杂交、磁珠富集及文库构建
PCR产物先95℃变性5 min后,与0.5 μmol/L生物素标记SSR探针 (AC)15和 (AG)15、4×SSC、0.1% SDS混合后在48℃杂交2 h。杂交液加至用TEN100(10 mmol/L Tris-HCl,1 mmol/L EDTA,100 mmol/L NaCl,pH值7.5)的平衡好1 mg链霉亲和素磁珠(New England Biolabs,美国)混合液中,室温恒温振荡温浴30 min,用磁架吸附管壁,将磁珠与混合液分离,弃混合液。然后依次用TEN1000(10 mmol/L Tris-HCl,1 mmol/L EDTA,1 mol/L NaCl,pH值7.5)漂洗2次,含0.1% SDS的0.2×SSC漂洗两次之后,加入100 μL TE重悬磁珠,95℃变性 5 min后,迅速用磁架吸附管壁将磁珠与混合液分离,保存上清液。以获得的上清液为模板用简并引物MseI-N进行PCR扩增,检测富集结果。PCR反应体系和程序与1.3.1中检测接头连接时相同。富集良好的PCR产物用Agarose Gel DNA Fragment Recovery Kit(TaKara,大连)回收纯化后,与pMD19-T载体(TaKara,大连)连接过夜,然后转化到大肠杆菌(E.coliDH5α)感受态细胞中,构建微卫星富集文库。
1.4 文库筛选、测序、序列分析及引物设计
将带有目的片段的大肠杆菌感受态细胞涂抹于含有Ampcillin (100 μg/mL)、X-gal(50 mg/mL)、IPTG(0.1 mol/L)的LB-琼脂平板培养基上,37℃过夜培养。挑取白色菌落于含Ampcillin的LB液体培养基中活化。以此活化菌液为模板,用载体通用引物和探针序列引物进行PCR扩增,筛选重组克隆。阳性克隆通过中国科学院高原生物适应与进化实验室ABI 3730xl型基因测序仪(Applied Biosystems,加拿大)进行DNA测序。
运用CLUSTAL X、MEGA和DNAsp对测定的序列进行分析,并加以手工校对[14-16]。利用SSRHunter软件对获得的序列进行分析[17],获得含有微卫星序列的片段。通过引物设计软件Primer3对获得的含有微卫星序列的片段进行引物设计[18]。
1.5 SSR标记引物多态性筛选
用高山绣线菊和蒙古绣线菊的4个野外居群(表1)进行SSR标记引物多态性筛选。PCR反应采用15 μL体系:10~100 ng DNA模板,1.5 μL的10×PCR Buffer(含1.5 mmol/L MgCl2),0.15 μL 10 mmol/L dNTP,正反引物各0.5 μL(5 pmol/L),0.5 UTaqDNA聚合酶(TaKara,大连),用ddH2O补足15 μL。PCR扩增反应程序:95℃预变性4 min;30个循环的94℃加热变性50 s,51℃~54℃(不同引物在不同物种中的退火温度不同,表2)退火45 s,72℃延伸30 s;最后72℃延伸7 min。PCR反应产物用QIAxcel Advanced 高效毛细管电泳仪(QIAGEN,德国)分析。
利用GENEPOP version 4.0.10计算每个位点的等位基因数量(number of alleles per locus)(A)、观察杂合度(observed heterozygosities;HO),期望杂合度(expected heterozygosities;HE)[19]。Hardy-Weinberg平衡检测用确切性概率方法(exact probability test)在GENEPOP中完成[19, 20]。
2 结果与分析
2.1 含目的片段阳性克隆筛选、测序及引物设计
以菌液为模板,用PCR法共筛选单克隆。产生两条或两条以上的扩增条带含有插入片段的阳性克隆262个(如图1中泳道1、2、3等),随机挑选其中120个进行测序。对获得的120条序列进行比对分析,结果显示其中111条序列(92.5%)为唯一序列,其他9条序列均为同一重复序列。SSRHunter软件对112条序列共检测到321个微卫星位点,平均每个克隆含有2.8个微卫星位点。挑选其中60条序列用引物设计软件Primer3设计出60对引物,由生工生物工程(上海)股份有限公司合成。通过优化PCR反应体系和程序,其中21对引物能在高山绣线菊和蒙古绣线菊中成功扩增,其中5对引物在初期筛选中未呈现出多态性,其余16对多态性引物的序列及GeneBank登记号在表2中详细列出。
2.2 多态性标记筛选
用获得的16对多态性引物(表2)对高山绣线菊和蒙古绣线菊的4个居群的92个个体进行PCR扩增,PCR产物用QIAxcel Advanced 高效毛细管电泳仪分析。图2所示为引物SA2对绣线菊的PCR扩增产物的毛细管电泳图谱。

表2 两种绣线菊部分有效扩增的微卫星引物
注:SA, 高山绣线菊Spiraeaalpina;SM, 蒙古绣线菊Spiraeamongolica。

图2 引物SA2部分PCR扩增产物QIAxcel Advanced 高效毛细管电泳图谱Fig 2 Profile of QIAxcel Advanced System with PCR production based on primer SA2
利用GENEPOP分析显示,高山绣线菊(P1和P2)中16个多态性位点的等位基因数(A)在3~18,观察杂合度介于0.043~0.870之间,期望杂合度的变化范围为0.126~0.950。在蒙古绣线菊中,这3个范围分别为:4~30、0.040~1.000、0.544~0.968。基于确切性概率方法的Hardy-Weinberg平衡检测发现,高山绣线菊和蒙古绣线菊的某些群体在多个位点显著偏离Hardy-Weinberg平衡(P<0.05),其中两个物种在SA2、SA6及SA10三个位点都发生了显著偏离(表3)。相比高山绣线菊,蒙古绣线菊的两个居群在11个位点都发生了显著偏离。
3 讨论
磁珠富集法使得SSR阳性克隆的获得率大大提高,传统的构建、筛选小插入片段基因组文库分离SSR遗传标记的方法中,阳性克隆的获得效率较低,只有1%~3%。多个研究表明磁珠富集法可以使得SSR阳性克隆的获得率提高至50%~60%[21]。本研究选取120个进行测序的克隆中,82个含有微卫星序列,获得率达到68.3%。近来通过高通量测序获得海量 EST序列(expressed sequence Tag),基于 EST序列大量开发 SSR 引物标记的方法正越来越引起人们的关注,然而由于高通量测序的成本较高,至少为磁珠富集法的5~8倍,其在研究中的应用还很有限。因此,作为一种快速,高效而廉价的微卫星标记分离方法,磁珠富集法在目前研究中得到广泛应用。此外,本研究用QIAxcel Advanced毛细管电泳代替了传统的变性聚丙烯酰胺凝胶电泳,该系统采用即用型的预制胶卡夹,能在最短的手工操作时间内分析96个样品,减少了人为操作误差,确保了便捷的数据分析和记录。
与多个其他类群的研究相比,绣线菊的4个居群的平均等位基因数、平均期望杂合度及平均观测杂合度都比较高,说明这16个位点在两种绣线菊中都具有较高的多态性,可以用于后续的遗传多样性、物种进化与亲缘关系等方面的研究[5]。微卫星多态性筛选中发现,64个数据系列(4个居群×16个位点)中26个显著偏离Hardy-Weinberg平衡。出现这种现象的可能是无效等位基因的存在。等位基因在理想群体中的频率是稳定的,无效等位基因的出现会引起等位基因频率的改变,从而导致绣线菊的居群在这些位点偏离Hardy-Weinberg平衡[22]。

表3 两种绣线菊居群遗传参数统计结果
注:*,显著偏离Hardy-Weinberg平衡。
有研究利用桉树随机基因组序列设计的SSR引物,在桉树属不同种间通用性约为78%[23],在桃金娘科不同属间仍具有30%以上的通用性[24]。利用玉米的SSR引物对棉花进行通用新分析发现,91%的引物能够成功扩增[25]。这是由于EST-SSR来源于编码区,因此在物种间具有很高的保守型。本研究是利用高山绣线菊的基因组DNA为材料进行SSR引物的开发,随机设计的60对引物中,21对引物能在高山绣线菊和蒙古绣线菊中成功扩增,其中16对在两个物种中均呈现较高的多态性。这说明在两种绣线菊中SSR引物的通用性也较高,本研究开发的16对SSR引物在绣线菊属的遗传学研究中具有较高的价值。在对绣线菊属的其他物种开展研究工作中,可以从已获得的高山绣线菊微卫星富集文库中寻找合适的微卫星位点,从而减少开发成本和提高实验效率。
近来,利用SSR标记进行的遗传多样性分析多有报道。通过筛选苜蓿的16对多态性丰富的SSR引物获得了190个多态性位点,分析发现这些SSR引物能够较好地检测不同苜蓿材料的遗传多样性[26]。利用自主开发的52对杉木SSR引物,对国家级杉木种质资源库保存的遗传材料分析表明SSR分子标记聚类的结果可为种质资源的管理及提高育种效率提供重要依据[27]。因此,本研究开发的SSR多态性引物将为绣线菊遗传多样性分析、物种进化与亲缘关系、种质资源利用、育种群体的建立等奠定基础。
参考文献:
[1]Tautz D. Hypervariabity of simple sequences as a general source for polymorphic DNA markers[J]. Nucleic Acids Research, 1989, 17(16): 6463-6471.
[2]Litt M, Luty J A. A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene[J]. American Journal of Human Genetics, 1989, 44(3): 397-401.
[3]Tautz D, Renz M. Simple sequences are ubiquitous repetitive components of eukaryotic genomes[J]. Nucleic Acids Research, 1984, 12(10): 4127-4138.
[4]Akagi H, Yokozeki Y, Inagaki A, et al. Microsatellite DNA markers for rice chromosomes[J]. Theoretical and Applied Genetics, 1996, 93(7): 1071-1077.
[5]姜立春, 彭正松, 阮期平. 微卫星 DNA 标记技术及其在植物研究中的应用[J]. 生物技术通讯, 2006, 17(4): 654-657.
[6]Rassmann K, Schlötterer C, Tautz D. Isolation of simple‐sequence loci for use in polymerase chain reaction‐based DNA fingerprinting[J]. Electrophoresis, 1991, 12(2/3): 113-118.
[7]Ostrander E A, Jong P M, Rine J, et al. Construction of small-insert genomic DNA libraries highly enriched for microsatellite repeat sequences[J]. Proceedings of the National Academy of Sciences, 1992, 89(8): 3419-3423.
[8]Ashizawa K, Kimura M K, Takahashi A, et al. Development of microsatellite markers in a riparian shrub,Spiraeathunbergii(Rosaceae)[J]. American Journal of Botany, 2012, 99(7): e283-e285.
[9]陆玲娣. 中国蔷薇科绣线菊亚科的演化, 分布——兼述世界绣线菊亚科植物的分布[J]. 植物分类学报, 1996, 34(4): 361-375.
[10]Lu L D, Gu C Z, Li C L, et al. Rosaceae[M].InWu Z Y, Raven P H, Hong D Y eds. Flora of China. Beijing: Science Press & Missouri: Missouri Botanical Garden Press, 2003, 46-434.
[11]Zhang F Q, Gao Q B, Zhang D J, et al. Phylogeography ofSpiraeaalpina(Rosaceae) in the Qinghai-Tibetan Plateau inferred from chloroplast DNA sequence variations[J]. Journal of Systematics and Evolution, 2012, 50(4): 276-283.
[12]张发起, 高庆波, 段义忠, 等. 横断山区高山绣线菊的谱系地理学研究[J]. 广西植物, 2012, 32(5): 617-623.
[13]Doyle J, Doyle J. A rapid DNA isolation procedure for small quantities of fresh leaf material[J]. Phytochemical Bulletin, 1987, 19:144-163.
[14]Thompson J D, Gibson T J, Plewniak F, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools[J]. Nucleic Acids Research, 1997, 25(24): 4876-4882.
[15]Tamura K, Stecher G, Peterson D, et al. MEGA6: molecular evolutionary genetics analysis version 6.0[J]. Molecular biology and evolution, 2013, 30(12): 2725-2729.
[16]Rozas J, Rozas R. DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data[J]. Computer Applications in the Biosciences: CABIOS, 1995, 11(6): 621-625.
[17]李 强,万建民. SSRHunter,一个本地化的SSR位点搜索软件的开发[J]. 遗传, 2005, 27(5): 808-810.
[18]Untergasser A, Cutcutache I, Koressaar T, et al. Primer3-new capabilities and interfaces[J]. Nucleic Acids Research, 2012, 40(15): e115-e115.
[19]Rousset F. genepop′007: a complete re-implementation of the genepop software for Windows and Linux[J]. Molecular Ecology Resources, 2008, 8(1): 103-106.
[20]Guo S W, Thompson E A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles[J]. Biometrics, 1992, 361-372.
[21]Zane L, Bargelloni L, Patarnello T. Strategies for microsatellite isolation: a review[J]. Molecular Ecology, 2002, 11(1): 1-16.
[22]Reece W H, Pinder M, Gothard P K, et al. A CD4+T-cell immune response to a conserved epitope in the circumsporozoiteproteincorrelates with protection from naturalPlasmodiumfalciparuminfection and disease[J]. Nature Medicine, 2004, 10(4): 406-410.
[23]Marques J F, Dehaene S. Developing intuition for prices in euros: rescaling or relearning prices?[J]. Journal of Experimental Psychology: Applied, 2004, 10(3): 148.
[24]Yasodha R, Ghosh M, Sumathi R, et al. Cross-species amplification of eucalyptus SSR markers inCasuarinaceae[J]. Acta Botanica Croatica, 2005, 64(1): 115-120.
[25]李华盛, 范术丽, 沈法富. 从棉花 ESTs 数据库中筛选微卫星标记的初步研究[J]. 棉花学报, 2005, 17(4): 211-216.
[26]陈 斐, 魏臻武, 李伟民, 等. 基于SSR 标记的苜蓿种质资源遗传多样性与群体结构分析[J].草地学报,2013, 21(4): 759-768.
[27]欧阳磊, 陈金慧, 郑仁华, 等. 杉木育种群体SSR分子标记遗传多样性分析[J]. 南京林业大学学报:自然科学版, 2014, 38(1):1-6.
猜你喜欢
特产研究(2022年6期)2023-01-17 05:05:06
农业开发与装备(2021年2期)2021-12-27 05:59:49
河北果树(2020年4期)2020-11-26 06:04:46
中国农业文摘-农业工程(2019年6期)2019-11-07 05:42:02
天然产物研究与开发(2018年10期)2018-11-06 07:43:50
四川动物(2017年4期)2017-07-31 23:54:19
中国医科大学学报(2015年10期)2015-03-01 02:09:55
集美大学学报(自然科学版)(2015年4期)2015-02-28 01:13:37
河北遥感(2014年3期)2014-07-10 13:16:48
天然产物研究与开发(2014年8期)2014-04-27 14:16:34
