
禾本科同源染色体对趋同进化的比较基因组学
2014-03-21 01:10马雪莲王振怡聂林曼汪厚龙王金朋
华北理工大学学报(自然科学版) 2014年2期
马雪莲,王振怡,聂林曼,汪厚龙,张 琼,王金朋
(河北联合大学生命科学学院,河北唐山063009)
0 引言
禾本科物种有着重要的经济和科学研究价值[1-3]。首先,它们是人类最主要的食物来源,如水稻(Oryza sativa)、玉米(Zeamays)和高粱(Sorghum bicolor)等为世界上大多数人提供主食;其次禾本科物种在进化上是成功的,在约五千到六千万年间,分化形成了700多个属,包含10000多个物种覆盖了地球20%的陆地面积。禾本科研究的主要动力无疑是其经济价值,而多个禾本科物种的全基因测序完成和即将完成为禾本科物种功能和进化的研究不断注入新动力[4-5]。
目前国内外关于禾本科植物全基因组加倍有了初步的研究和探索,有研究表明禾本科物种的共同的祖先物种在七千万年前发生了一次全基因组的加倍(Whole gene duplication)[3,6-7],全基因组加倍之后物种基因组中产生大量重复基因,为物种基因组的遗传创新提供了丰富的材料来源。遗传创新的一种重要机制就是同源DNA片段的遗传重组(Genetic recombination),不仅可以对遗传过程中的DNA序列的有害变异进行修复,而且可以进行同源序列间信息传递[8-9],即同源重组。相对于同源重组非同源染色体对间的重组称为非正常遗传重组(illegitimate recombination)[10]。多倍化之后物种基因组极为不稳定,出现DNA丢失,倒位和染色体重排[3,11]。基因组重排的结果就是有新的同源染色体对(Neo-homologous chromosomes)产生[10],这一新同源染色体对相对于基因组重排之前并非同源染色体对,而是全基因组加倍之后产生,那么基因组的这一重排机制在一定程度上可以抑制染色体重组,然而这一同源染色体对在物种基因组中是否独立进化并保持下来,是一个十分值得探索的问题。
最近,禾本科植物水稻、高粱、玉米、谷子(Setaria italica)和二叶短柄草(Brachypodium distachyon)的全基因组测序率先完成[5,12-16],为比较基因组学研究提供了良好的数据材料。本文以五个禾本科物种全基因组序列为研究对象,对其基因组进行同源性和染色体结构的比较分析,确定物种基因组内由全基因组加倍产生的重复基因片段,并建立基因组间染色体片段的同源信息,通过种内和种间重复基因比较推断七千万年来同源染色体对趋同进化的规律,揭示物种分化演化的可能驱动力,加深DNA重排和非正常遗传重组对基因组结构进化影响的认识。
1 材料和方法
1.1 数据材料来源
研究所需的5个禾本科植物水稻、高粱、玉米、谷子和二叶短柄草的全基因组序列从公共数据库上下载得到:植物全基因组加倍数据库http://chibba.agtec.uga.edu/和植物基因组数据库http://www.jgi.doe.gov/。下载获得每个物种的DNA序列,蛋白质序列以及基因组的注释信息。
1.2 基因组同源性分析
利用序列同源搜索工具Blastp分别对谷子和谷子,玉米和玉米,谷子和玉米的蛋白序列进行比对分析,确定基因间的同源性(E-value<1e-5);根据获得的基因同源性以及基因在染色体上的物理位置画二维平面图,图中每个点表示一同源基因对,要求基因对匹配的打分值大于100,对于每个基因去掉保留前5个匹配基因对,最好匹配点为红色,次好匹配为蓝色,其它为灰色。
1.3 推断基因共线性
首先,利用多重序列比对工具McScan[17]寻找基因组内和基因组间同源共线DNA片段;然后,利用加入统计估计的共线性分析工具ColinearScan[18],对获得的基因组共线区域进行统计估计其显著性,将显著性大于1e-10的区域去掉,确定基因组内重复基因和基因组间的同源共线信息;最后,根据上述同源性信息以及基因组结构分析获得物种多重序列比对结果。
1.4 分子进化距离估计
同源基因对间的分子进化距离,用核苷酸置换率Ks进行度量,利用物种进化分析软件PAML[23]中的包含的Nei-Gojobori方法计算[24]。
2 结果及讨论
2.1 禾本科植物种内基因组同源结构
比对分析物种基因组获得的基因组内的同源信息,根据得到的基因同源信息,染色体的长度以及基因在染色体上的物理位置,对每个物种以及两物种之间的基因组结构进行分析,以水稻和玉米为例展示研究结果,结果是以基因结构点阵图的形式给出(如图1)。

图1 图为水稻和玉米基因结构点阵图
图1图为水稻和玉米基因结构点阵图(Blastp比对基因组蛋白序列):二维平面中的横轴从左向右依次分别排列的是水稻的12条,玉米10条染色体,纵轴从上到下依次排列,图中每个点表示的是一对同源基因(基于氨基酸序列相似性获得),最好匹配的点为红色,次好匹配为蓝色,其它为灰色。在图中能够观察到一系列由连续的点构成的线,这些线上的基因对是由基因倍增产生的大量重复基因。
图中可以看到每个物种基因组中有大量的共线性同源基因片段存在,并且相对于水稻基因组玉米中有更多的共线性片段。在水稻基因组中一些共线性基因片段分别存在于Os01-Os05,Os02-Os04,Os02- Os06,Os03-Os07,Os08-Os09,Os11-Os12;在玉米基因组中Zm01-Zm05,Zm01-Zm09,Zm02-Zm07,Zm02-Zm10,Zm03-Zm08,Zm04-Zm05,Zm06-Zm08,Zm06-Zm09。
上述共线区域分别存在两物种基因组中的重复基因,这些重复基因由全基因组加倍产生,但在物种进化过程中保留了较好的共线性片段。如水稻的1和5号染色体是由它们共同祖先物种的同一染色体加倍而来,从染色体上基因间的同源性可以判断这两条染色体序列几乎一致,但仍有一些基因序列并非同源基因,这可能是由于物种基因组加倍之后,重复基因进化导致基因序列间的差异变大的结果;相对于谷子基因组玉米有更多的同源染色体片段,且染色体片段较短,在点图中呈现蓝色的线,与红色的线相比相似性差一些,这些重复基因是由和谷子共同加倍产生[3],但在后来的进化过程中基因序列变异导致相似性降低,并且伴随大量的基因丢失[11],较长的由红色点构成的染色体片段是由较近的基因加倍产生[19]。这里以水稻和玉米展示了其基因组同源结构,相应的规律同样存在于高粱、谷子以及二叶短柄草。
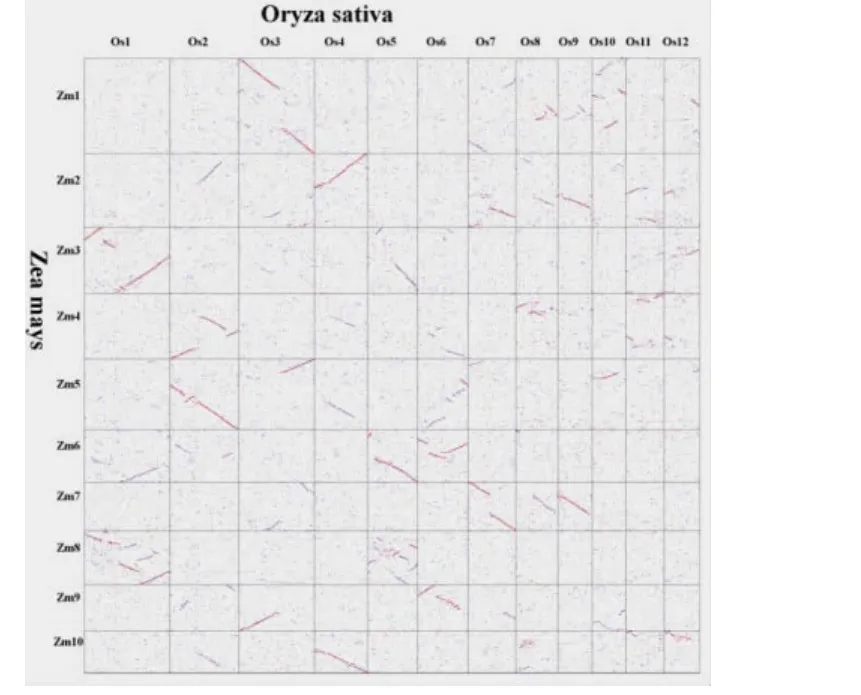
图2 水稻和玉米之间基因结构点阵图
2.2 禾本科植物种间基因组同源结构
种间基因组比较,实现同源染色体片段分组,确定物种间真正直系同源基因。这一过程分析了谷子和玉米物种间基因组点阵图,并结合同源基因相似性,理清不同进化事件产生的同源片段,并把不同的同源组分组。在图中可以发现水稻每条染色体都会对应两条比较好的染色体片段,如玉米的1号染色体对应最好的玉米的两条染色体分别是3和8号染色体,它们之间的同源染色体片段在图中形成了两条红色的线,而谷子的5号和1号染色体是一对旁系同源基因对,并且这一规律在其它染色体上也都存在,这一结果表明玉米在其进化过程中不仅与水稻共同发生过一次古老的全基因组加倍,在之后发生过再次的全基因组加倍[19]。同源基因对在图中形成的红色的线表示两物种之间真正的直系同源基因片段,如水稻1号和玉米的3号染色体上有3个直系同源染色体片段;但同一染色体区域上的蓝色线是由种外的旁系同源基因构成,如水稻的1号和玉米的6号染色体之间的同源染色体片段。通过上述分析水稻与其它4个禾本科物种,可获得水稻与其它每个物种间的基因同源关系。
图2.点阵图展示了基因组间的序列比对信息。红色的线是Blast比对两物种基因组序列获得的最好匹配的基因对,表示物种间直系同源染色体片段;蓝色是次好匹配构成的线,表示种间旁系同源染色体片段。
2.3 种内及种间同源染色体片段
基于前面基因同源信息,以及基因组结构分析,这里获得了所有物种内以及物种间的同源染色体片段。如高粱基因组内同源染色体片段(如图1a):Sb01-Sb01,Sb01-Sb02,Sb01-Sb08;Sb02-Sb07;Sb03-Sb09;Sb04-Sb06,Sb04-Sb10;Sb05-Sb08;Sb06-Sb07(注:Sb表示高粱,如Sb01表示高粱1号染色体);玉米染色体内同源染色体片段(如图1b):Zm01-Zm05,Zm01-Zm07,Zm01-Zm09;Zm02-Zm04,Zm02-Zm05,Zm02-Zm07,Zm02-Zm,10;Zm03-Zm06,Zm03-Zm08,Zm03-Zm10;Zm04-Zm05,Zm04-Zm09,Zm04-Zm,10;Zm05-Zm06,Zm05-Zm09,Zm05-Zm10;Zm06-Zm08,Zm06-Zm09;高粱和玉米之间同源染色体片段(如图2):
Sb01-Zm01,Sb01-Zm05,Sb01-Zm09;Sb02-Zm02,Sb02-Zm07;Sb03-Zm03,Sb03-Zm08;Sb04-Zm04,Sb04-Zm05;Sb05-Zm02,Sb05-Zm04,Sb06-Zm02,Sb06-Zm10;Sb07-Zm01,Sb07-Zm04,Sb07-Zm10;Sb08-Zm01,Sb08-Zm03,Sb08-Zm10;Sb09-Zm06,Sb09-Zm08;Sb10-Zm05,Sb10-Zm06,Sb10-Zm09。
2.4 禾本科植物基因组中特殊同源染色体对
水稻11和12号染色体是由全基因组加倍产生的一对部分同源染色体对,该染色体对上保留了大量的重复基因,遗传过程中这一基因对独立保留下来同时进行着相互作用[10]。根据上述染色体结构分析以及物种染色体片段间的同源关系,这里确定了物种部分同源染色体对,如水稻11号12染色体与其它物种之间的同源染色体对,水稻和高粱:Os11-Sb05;Os12-Sb08;水稻和玉米:Os11-Zm02;Os12-Zm01,Os12-Zm10;水稻和谷子:Os11-Si08;Os12-Si03,Os12-Si09;水稻和二叶短柄草:Os11-Bd04(11号对4号中间部分); Os12-Bd04(12号对4号上下部分)。
2.5 禾本科植物基因组中的共线性分析
基于基因同源共线性分析获得了每个禾本科物种基因组中含有全基因组加倍产生的重复基因的对数超过2700对,占物种全基因组的15%以上,且重复基因常常是分布在靠近染色体末端的位置(如图3)谷子中的重复基因。在谷子基因组内共有402个共线性区域,包含3846对旁系同源基因对,其中最长的区域存在与3号与5号染色体之间,有279对旁系同源基因对,长度大于10对旁系同源基因对的区域有60个,大于50对的区域有8个;谷子全基因组有约40000个基因,其中重复基因占全部基因的19.2%。

图4 (b)水稻11和12号染色体同源染色体进化模式
图4 (a)谷子中的重复基因
图4.水稻11和12号染色体与高粱5号和8号,谷子7号,8号和3号染色体之间的同源模式及进化模式以及他们的共同祖先:染色体片段(RSA-RSC,SSA-SSB和CSA-CSC),RS表示水稻-高粱分化之后,水稻11号和12号上的染色体片段,SS表示高粱5号和8号上的染色体片段;CS表示在两物种分化之前水稻-高粱共同的染色体片段。水稻12号和高粱8号染色体上的特有片段用ADD表示。染色体上红色的曲线表示每条染色体上基因的密度,S和L分别表示染色体短臂、长臂,圈中曲线连接的是重复基因对,基因对之间曲线的颜色根据分子距离渐变。
2.6 同源染色体对趋同进化的初步推断
为了推断同源染色体对间趋同进化,这里计算了物种同源染色体对上重复基因的分子距离,并且进行了多次的统计实验分析,研究的结果(如图4),分析发现水稻的11号和12号染色体是一对同源染色体对,对应高粱的5号和8号染色体,以及谷子的3号,7号和8号染色体,圈中不同颜色的曲线代表了重复基因对的分子距离的不同,观察可以发现靠近染色体短臂的重复基因对间的分子距离特别小,这表明在基因组进化过程中这些基因对可能发生了基因置换,也即同源的染色体对间存在趋同进化;另外可以看到自染色体短臂到染色体长臂,重复基因对之间的距离有渐变的过程,这表明同源染色体对间的趋同进化可能受重排抑制,并呈阶段的发生。值得注意的是在谷子基因组进化过程中其3号和7号染色体有共同的祖先染色体,它们在进化过程中发生过染色体断裂事件,并且这一祖先染色体序列的一个片段与3号染色体融合,另一片段与7号染色体融合为两条新的染色体序列;从图中水稻-高粱的同源染色体对上可以看到在靠近着丝粒区几乎不存在重复基因。
3 结论
禾本科物种自七千万年前的多倍化后,基因组结构的复杂性增加,为揭示非正常遗传重组对基因结构的影响,本文对五个禾本科物种的全基因组进行了比较分析。统计推断了物种基因组内由全基因组加倍产生的重复基因片段,并确定了种内和种间的同源染色体片段;基于基因序列相似性比较,发现同源染色体对上越靠近染色体末端的重复基因间的分子距离越,这可能是重复基因间的非正常遗传重组导致同源染色体对间的DNA序列变得更为相似的原因。然而关于非正常遗传重组发生的模式,以及如何影响物种基因组进化,有待于进一步深入研究。
[1] Kellogg,E.A.Relationships of cereal crops and other grasses.Proceedings of the National Academy of Sciences of the United States of America,95 (5):2005-2010,1998.
[2] Gaut,B.S.Evolutionary dynamics of grass genomes.New Phytologist,154:15-28,2002.
[3] Paterson,A.H.,Bowers,J.E.,and Chapman,B.A.Ancient polyploidization predating divergence of the cereals,and its consequences for comparative genomics.Proc Natl Acad Sci USA,101(26):9903-9908,2004.
[4] Yu,J.Wang,J.Lin,W.Li,S.G.Li,H.Zhou,J.Ni,P.X.Dong,W.Hu.S.N.Zeng,C.Q.et al.The Genomes of Oryza sativa:A history of duplications.PLOSbiology,3(2):266-281,2005.
[5] Paterson,A.H.,Bowers,J.E.,Bruggmann,R.,Dubchak,I.,Grimwood,J.,Gundlach,H.,Haberer,G,Hellsten,U.,Mitros,T.,Poliakov,A.etal.The Sorghum bicolor genome and the diversification of grasses.Nature,457(7229):551-556,2009a.
[6] Kellogg,E.A.Relationships of cereal crops and other grasses.Proceedings of the National Academy of Sciences of the United States of America,95 (5):2005-2010,1998.
[7] Gaut,B.S.Evolutionary dynamics of grass genomes.New Phytologist154:15-28,2002.
[8] Puchta H.,Dujon B.,and Hohn B.,etal.Two differentbut relatedmechanisms are used in plants for the repair ofgenomic double-strand breaks by homologous recombination[C].Proc.Natl.Acad.Sci.93:5055-5060,1996.
[9] Khakhlova O.,and Bock R..Elimination of deleteriousmutations in plastid genomes by gene conversion[J].Plant J.,46:85-94,2006.
[10] Wang X.,Tang H.,et al.Comparative inference of illegitimate recombination between rice and sorghum duplicated genes produced by polyploidization[J].Genome Res,19(6):1026-32,2009.
[11] Wang,X.,X.Shi,et al.Duplication and DNA segmental loss in the rice genome:implications for diploidization[J].New Phytol,165(3):937-46,2005.
[12] International Rice Genome Sequence Project.Themap-based sequence of the rice.Nature,Vol.436,doi:10.1038,793-800,2005.
[13] The International Brachypodium Initiative.Genome sequence and analysis of themodel grass Brachypodium distachyon.Nature,vol.463,doi:10.1038,763-768,2010.
[14] Zhang G.Y.,Liu X.,Wang J.,etal.Genome sequenceof foxtailmillet(Setaria italica)provides insights intograssevolution and biofuel potential[J].Nature Biotechnology,vol.30,no.6,pp.549-556,2012.
[15] Bennetzen J.L.,Schmutz J.,Devos K.M.,et al.Reference genome sequence of themodel plant Setaria[J].Nature Biotechnology,vol.30,no.6,pp.555-561,2012.
[16] Schnable P.S.et al.The B73 maize genome:complexity,diverisity and dynamics[J].Science,326,pp.1112-1115,2009.
[17] Tang H.,Wang X.,Bowers J.E.,et al.Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps[J].Genome Res,18: 1944-1954,2008.
[18] Wang X.,Shi X.L.,Li Z.,etal.Statistical inference of chromosomal homology based on gene colinearity and applications to Arabidopsis and rice[J].BMC Bioinformatics,7:447,2006.
[19] J.C.Schnable,N.M.Springer,M.Freeling.“Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss”.PNAS,vol.108,no.10,pp.4069-4074,2011.
猜你喜欢
当代陕西(2021年21期)2022-01-19
汉字汉语研究(2021年2期)2021-08-30
汉字汉语研究(2019年2期)2019-08-27
新高考·英语进阶(高二高三)(2018年8期)2018-01-15
中国中药杂志(2017年17期)2017-09-23
河北书画研究(2016年3期)2016-04-28
湖南城市学院学报(自然科学版)(2016年4期)2016-02-27
农产品市场周刊(2015年39期)2015-10-31
现代农业(2015年5期)2015-02-28
现代农业科技(2009年9期)2009-02-21
