
光催化合成金属Ag纳米颗粒的生长机制——晶核密度控制的生长模式转换
2014-01-18 06:16张庆瑜
无机化学学报 2014年7期
李 帅 陶 强 张庆瑜
(大连理工大学三束材料改性教育部重点实验室,大连 116024)
在光的作用下,贵金属纳米颗粒,如Au、Ag纳米颗粒,可以形成等离子激元(plasmon)共振效应,使其在催化、生物传感、光电子学和表面增强拉曼光谱(SERS)等众多领域有着广泛的应用[1-9]。因为金属纳米颗粒的性质与其尺寸、形状和结构有关[10-13],因此控制纳米颗粒的生长,从而形成各种各向异性的金属纳米颗粒是开展各种应用研究的基础[14-15]。例如,在基于等离子体激元增强光催化与光伏方面的应用中,各向异性的金属纳米颗粒相对于各项同性的纳米颗粒通常在可见和红外光区域有更好的等离子体激元消光特性,这种特性将有助于光催化与光伏材料的光吸收性能向可见和红外区域扩展,进而增强其在太阳光下的性能[16-18]。
在各种金属纳米颗粒的合成方法中,化学反应合成是制备金属纳米颗粒的主要方法。通过使用表面覆盖剂,如聚乙烯吡咯烷酮(polyvinyl pyrrolidone,PVP),纳米颗粒的生长动力学过程可以通过温度控制向完全不同的各向异性纳米结构方向进行[19-21]。而且,与传统的Ostwald熟化机制(Ostwald Ripening,OR)不同,人们提出了一种以微小颗粒为基本单元,进而自组装生长成纳米颗粒的取向附生(oriented attachment,OA)解释纳米颗粒的各向异性生长行为[22-26]。但是,化学合成方法中表面覆盖剂和活性剂往往会残留在纳米金属颗粒的表面,从而产生一些副作用。例如,SERS是一种微量检测技术,甚至可以实现有机分子的单分子检测,而残留在金属纳米颗粒表面的有机分子将产生背景信号干扰,从而引起SERS检测灵敏度的下降。
光催化合成是制备金属纳米颗粒的一种有效手段。与化学反应合成不同,光催化合成是通过光生载流子还原溶液中的金属离子实现纳米金属颗粒的生长,从而有效避免了表面活性剂等有机分子的污染。利用AgNO3水溶液,光催化方法可以在一些半导体表面,如TiO2薄膜,制备出金属Ag纳米颗粒[27-30]。但是,由于缺少表面覆盖剂的辅助,生长出的Ag纳米晶粒往往是不规则的球形,这对调控纳米金属颗粒的光学性质是非常不利的。因此,控制金属颗粒实现各向异性生长是光催化合成中的一个重要课题。最近,Tanabe等通过光催化方法,利用AgNO3水-乙醇溶液合成了金属Ag纳米片[29]。随后,Wang等人报道了具有规则多面体形状和树枝状的Ag纳米颗粒的光催化合成[27]。但是,有关光催化合成中金属Ag纳米颗粒的各向异性生长机制并不十分清楚。
本文利用AgNO3水溶液,通过严格控制TiO2薄膜的化学活性,系统研究了光催化合成金属Ag纳米颗粒的生长行为。研究结果显示,光催化合成金属Ag纳米颗粒存在着两个完全不同的生长机制,即OR机制和OA机制,分别对应着金属Ag纳米颗粒的各向同性和各向异性生长。AgNO3溶液的浓度,更准确地说是Ag初级晶核的局域密度是控制纳米颗粒生长动力学行为的重要因素。在此基础上,本文提出了有关光催化生长金属Ag纳米颗粒的物理模型。该模型或可为合成各向异性Ag纳米颗粒及其生长机理的解释提供新的思路,同时这种简易的直接在TiO2表面合成Ag纳米片的方法有望在等离子激元增强光催化与光伏,生物传感,表面增强拉曼与荧光等方面有潜在的应用。
1 实验方法
1.1 溶胶凝胶TiO2薄膜的制备
利用溶胶凝胶方法来制备光还原沉积Ag纳米颗粒所用的TiO2薄膜。首先,量取50 mL的钛酸丁酯和3 mL的乙酰丙酮混合于烧杯中并于磁力搅拌器上搅拌,再将0.21 mL是硝酸,1.4 mL的去离子水和150 mL的无水乙醇混合充分搅拌后配制成滴加液,将滴加液缓慢滴加入正在强烈搅拌的钛酸丁酯和乙酰丙酮混合溶液,再搅拌0.5 h后,放置1 d时间陈化待用。将清洗干净的玻璃基片浸渍于配好的TiO2溶胶后提拉成湿膜,湿膜于550℃下退火2小时即得到结晶的TiO2薄膜。
1.2 光催化生长Ag纳米颗粒
将同一次制备的带有TiO2薄膜的载波片切割成小块用于金属Ag纳米颗粒的生长,以确保金属Ag纳米颗粒生长条件的一致性。金属Ag纳米颗粒的生长是在暗室中进行的,以避免杂散光对光催化过程的影响。将涂有TiO2薄膜的玻璃基片浸没在AgNO3水溶液中,膜面朝上。采用低压汞灯(Philips TUV,8 W)为辐照光源。为了研究金属Ag纳米颗粒的生长行为,利用紫外-可见-近红外光纤光谱仪(Ocean Maya 2000-Pro)原位检测生长过程。原位光谱采集间隔为10 s,波长范围为200~1 100 nm。光照停止以后,将样品从溶液池中取出,并用干燥氮气吹干。利用扫描电子显微镜(SEM,S-4800)分析金属Ag纳米颗粒的形貌。利用透射电子显微镜(TEM,JEM-2000EX)分析Ag纳米片的结构。
2 结果与讨论
2.1 金属Ag纳米颗粒随时间的演化
图1是AgNO3溶液浓度为180 mg·L-1时金属Ag纳米颗粒的形貌随时间的演化。可以看到,金属Ag纳米颗粒呈不规则的球形,晶粒尺寸随生长时间的增加而增大,说明金属Ag纳米颗粒以各向同性的模式生长。我们注意到,在生长的初期,如图1(a)所示,金属Ag纳米颗粒有两种完全不同的晶粒尺寸,其中尺寸较小的晶粒具有非常高的密度。当生长时间超过4 min以后,这种小尺寸晶粒的密度骤然下降,同时尺寸较大的晶粒呈现出明显的OR生长特征,如图1(b)~(d)所示,说明此时的金属Ag纳米颗粒属于OR机制主导的生长。图1(c)中插图为Ag纳米颗粒8 min样品的XRD结果,图中的38°和44°分别对应面心立方结构的Ag晶体的(111)与(200)面。
图2是AgNO3溶液浓度为 2 800 mg·L-1时金属Ag纳米颗粒的形貌随时间的演化。与低溶液浓度时的生长完全不同,此时的金属Ag纳米颗粒呈现出片状形貌。金属Ag纳米片以三角形为主,此外还有一些不规则的多边形。从SEM照片中可以明显看出,这些金属Ag纳米片非常薄,下面的金属Ag颗粒依稀可见,说明纳米片的厚度非常薄。根据垂直生长的Ag纳米片,可以估算出Ag纳米片的厚度在10~20 nm之间,而纳米片的尺寸在几百纳米到几微米量级,说明此时金属Ag纳米颗粒表现出非常强烈的各向异性生长。图1(a)中插图为将样品超声后获得的单个的Ag纳米片的TEM图像以及其电子衍射图。图中衍射斑点的形状表明Ag纳米片的表面为(111)晶面。
令人吃惊的是,光照时间仅为2 min时,Ag纳米片就已经形成,但Ag纳米片的尺寸差异较大,最大Ag纳米片的边长达到微米量级,如图2(a)所示,远远大于低浓度溶液(180 mg·L-1)时的晶粒尺寸。随着光照时间的增加,小尺寸Ag纳米片的数量明显减少,而大尺寸Ag纳米片的数量有所增加,如图2(b)和2(c)所示。从形貌上看,此时三角形Ag纳米片的比例有所下降,而不规则多边形,如五边形、六边形以及锯齿形纳米片的比例明显增加,这意味着这些较大尺寸的多边形可能是通过小尺寸的三角形纳米片以OA机制合并而成的。从更高倍数的SEM电镜图片(图3(a))中可以更明显的看到片与片之间相互连接合并的分界线以及一些三角片相互合并成锯齿形状的Ag纳米结构,如图3(a)中圆圈所显示。利用TEM可以观察到片与片的相合并过程中留下了明显的晶界,如图3(c)所示。高分辨TEM(图3(d))显示晶界两边具有着近乎一致取向的Ag(111)晶面。这些观察说明片与片之间合并是在一定的取向上来进行的。再仔细观察在一些Ag纳米片的表面并不完全的平整,而是存在一些坑洼的缺陷,如图3(a)中方框中所示,这可以认为是颗粒在以OA机制合并未完全所留下来的。而我们确实也在一些Ag纳米片的表面发现了更为明显的颗粒构成片的结构,如图3(b)中所示。上述实验现象充分的证明了我们所制备的Ag纳米片的生长过程是一个由OA机制所主导的由小的初级颗粒合并为纳米片,再由小的纳米片逐渐合并成大的纳米片的过程。

图1 180 mg·L-1AgNO3浓度下不同光催化生长时间的Ag纳米颗粒的扫描电镜图Fig.1 Typical SEM images of Ag nanoparticles grown in 180 mg·L-1AgNO3solution for different irradiation time:(a)2 min,(b)4 min,(c)8 min,(d)16 min

图2 2 800 mg·L-1AgNO3浓度下不同光催化生长时间的Ag纳米颗粒的扫描电镜图Fig.2 Typical SEM images of Ag nanoparticles grown in 2 800 mg·L-1AgNO3solution for different irradiation time:(a)2 min,(b)4 min,(c)8 min,(d)16 min

图3 (a),(b)更高倍数的Ag纳米片的扫描电镜图;(c),(d)Ag纳米片相互合并位置的透射电镜图片Fig.3 (a),(b)SEM images of Ag nanoplates;(c),(d)TEM and HRTEM images of Ag nanoplates

图4 生长时间为2 h的Ag颗粒形貌Fig.4 Typical SEM images of Ag nanoparticles grown in 180 mg·L-1(a)and 2 800 mg·L-1(b)AgNO3solution for 2 h
当光照时间达到16 min时,金属Ag纳米片的厚度明显增加,但尺寸有所减小。同时,出现了一些比较规则的多面体Ag纳米晶粒,如图2(d)所示。我们推测,随着金属Ag纳米片尺寸和厚度的增加,纳米片内部的应力逐渐增大。当内应力增加到足够大时,大尺寸纳米片将分裂成小片,这可能是Ag纳米尺寸减小的主要原因。进一步延长光催化生长时间发现,光催化产物与低溶液浓度的结果类似,但片状Ag颗粒的比例相对较大,如图4所示,说明金属Ag纳米片的生长后期确实存在一个分裂过程,进而转变为以OR机制生长为主。
2.2 金属Ag纳米颗粒的消光光谱分析
图5是金属Ag纳米颗粒的消光光谱随时间的变化。从图中可以看出,各向同性生长的金属Ag纳米颗粒的透射光谱中呈现出明显的球状金属颗粒消光特征,如图5(a)所示,其中370 nm附近的消光峰对应着金属Ag纳米颗粒中局域表面等离子激元(LSP)的四极矩共振消光,450~550 nm的长波消光峰是LSP的偶极矩共振消光[31]。当金属颗粒尺寸较小时,偶极共振消光占主导,随着晶粒尺寸的长大,四极共振逐渐增强,这与SEM形貌观察的结果是一致的。同时,随着单位面积上金属Ag质量密度的增加,长波波段(1 000~1 100 nm)的背景消光逐渐增强,这是LSP有效介质密度变化的结果[32-33]。与各向同性生长的金属Ag纳米颗粒不同,金属Ag纳米片的消光光谱在370~1 100 nm的波长范围内均比较平坦,如图5(b)所示,更接近金属Ag薄膜的消光光谱。但辐照时间达到8 min以后,消光光谱中出现了金属Ag纳米颗粒的消光趋势,370~600 nm波段的消光逐渐增强,这可能与Ag纳米片分裂成小片有关。
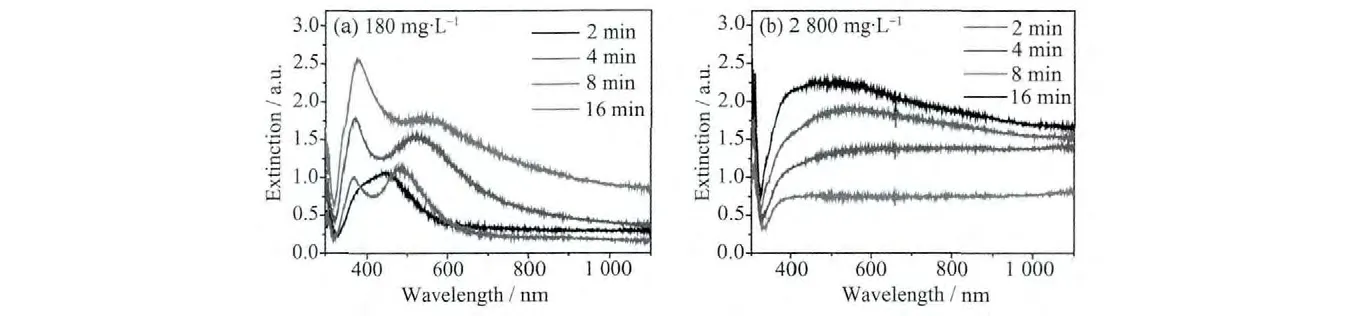
图5 不同生长时间的Ag纳米颗粒的消光光谱Fig.5 Extinction spectra of Ag nanoparticles in 180 mg·L-1(a)and 2 800 mg·L-1(b)AgNO3 solution for different irradiation time

图6 Ag纳米颗粒的原位实时消光光谱Fig.6 In situ and real-time extinction spectra of Ag nanoparticles in 180 mg·L-1(a)and 2 800 mg·L-1(b)AgNO3solution
由于SEM形貌观察的非原位特征以及分辨率的限制,无法获得更短时间尺度上有关金属Ag纳米颗粒生长的可靠信息。为了进一步了解两种不同生长模式的金属Ag纳米颗粒的生长行为,我们对比了它们生长初期的原位消光光谱,如图6所示,其中消光光谱的数据采集间隔为10 s。由于原位消光光谱的采集是在水溶液环境下进行的,与图5中的空气环境有所不同,消光光谱的形状发生了一些变化。不过,这两种生长模式的差异仍十分明显。从图中可以看到,当光照时间仅为10 s时,就已经出现了明显的金属Ag颗粒的LSP消光。为了避免概念的混淆,我们定义初期生长的小尺寸纳米颗粒为晶核。在低浓度溶液中,消光峰有2个,分别位于380 nm和500 nm,说明消光来源于球状金属Ag晶核,由于部分晶核尺寸较大,出现了四极共振消光;在高溶液浓度中,尽管消光光谱比较平坦,在500 nm附近仍可以看到1个比较弱的消光峰,但观察不到380 nm附近的肩峰,说明此时溶液中的金属Ag晶核也是球状的,但尺寸相对较小,密度较大。随着光照时间的增加,低浓度溶液一直保持着类球状的消光特征,但LSP的四极共振消光的贡献逐渐增强,说明晶核以各向同性的模式长大成金属Ag纳米颗粒。然而,在高浓度溶液中,10~30 s的消光光谱发生了较大变化,长波波段(800~1 100 nm)的消光急剧增强,同时在330 nm附近出现了1个小的肩峰,如图中箭头所示。大量实验结果显示,AgNO3水溶液中340 nm出消光峰与三角片状金属Ag纳米片的LSP面外(out-of-plane)四极共振有关[31]。同时,三角片状金属Ag纳米片的LSP面内(in-plane)偶极共振将随着边长的增加而向长波方向移动[26],这应该是长波波段消光急剧增加的主要原因。上述分析表明,在高浓度溶液中,存在着一个从晶核到金属Ag纳米片的转变,这种转变显然不属于OR生长机制,而应该与OA生长机制有关。
2.3 光催化制备Ag纳米颗粒的生长机制
上述研究结果表明,AgNO3溶液的浓度是控制金属Ag纳米颗粒生长模式重要因素。不同AgNO3浓度下的实时消光光谱的研究结果表明(图7),对于特定的TiO2薄膜,控制金属Ag纳米颗粒形貌的转变存在一个临界浓度,介于300~400 mg·L-1之间。当AgNO3溶液浓度小于300 mg·L-1时,光催化合成产物呈现各向同性生长特征,为不规则的金属Ag纳米颗粒;当AgNO3溶液浓度大于400 mg·L-1时,光催化合成产物呈现各向异性生长特征,为金属Ag纳米片。这一结果与化学反应合成金属纳米颗粒有着本质的不同。在化学反应合成中,一般认为,表面覆盖剂对金属Ag纳米颗粒的各向异性生长起着至关重要的作用,表面覆盖剂通过抑制某些晶面的生长或取向附生而实现各向异性生长。然而,在我们的光催化合成过程中,没有人为添加的表面覆盖剂及其它有机分子,金属Ag纳米颗粒的生长模式完全通过溶液浓度控制。同时,我们的研究结果表明,各向同性生长的金属Ag纳米颗粒为OR机制主导的生长过程,而各向异性生长的金属Ag纳米片为OA机制主导的生长过程。决定光催化产物以OR机制生长还是以OA机制生长的关键是AgNO3溶液溶度,或更准确地说是初期晶核的尺寸和密度。因此,一定存在一个与化学反应合成不同的各向异性生长控制机制。
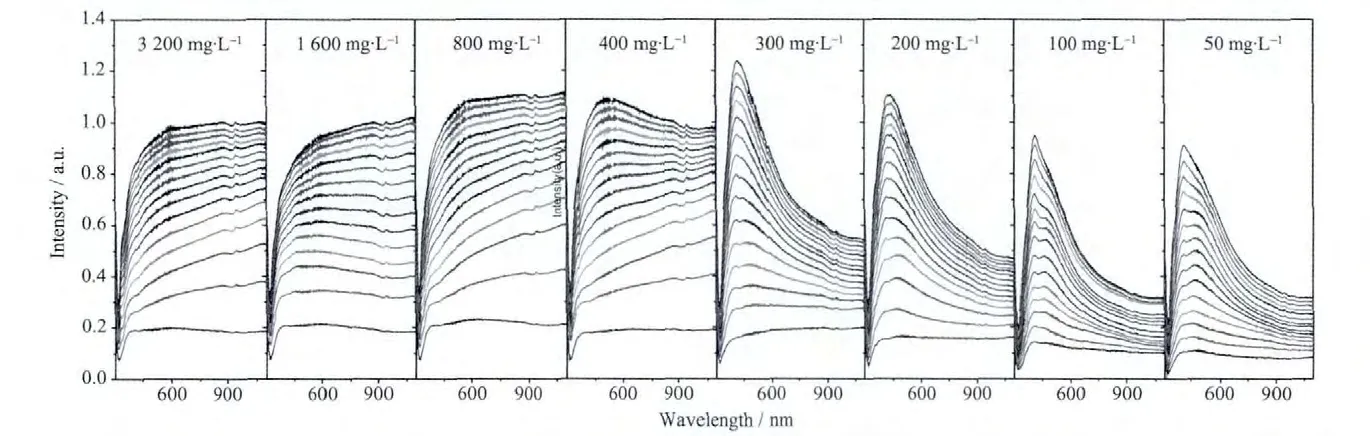
图7 不同AgNO3浓度下光催化生长的Ag纳米颗粒的原位实时消光光谱Fig.7 In situ and real-time extinction spectra of Ag nanoparticles in AgNO3solutions with different concentration
晶体生长理论表明,晶体的成核自由能与溶液中离子的过饱和度有关。对于自发成核过程,成核自由能为[34]
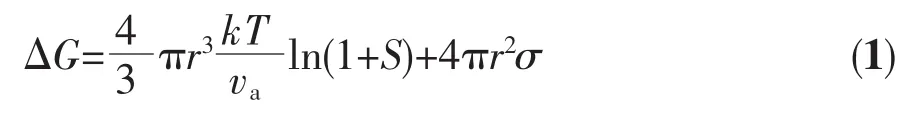
其中,va是原子或分子的体积,r是晶核半径,k是玻尔兹曼常数,σ是晶核表面自由能密度,S=C/C0-1是过饱和度,C和C0分别是原子和分子的过饱和浓度和饱和浓度。临界成核自由能、临界晶核半径及临界晶核密度分别为[31]

可以看到,过饱和度越高,晶核的形成自由能越低,成核越容易,晶核的尺寸越小,晶核密度越大。异质成核与自发成核类似,但受基片的影响,成核自由能更低,与接触角有关。
在光催化合成金属Ag纳米颗粒的过程中,Ag原子是通过溶液中Ag+离子与TiO2薄膜表面产生的光生载流子(电子)复合而形成的。因此,Ag原子的产生率正比于光生载流子的产生率和溶液中TiO2薄膜表面附近的Ag+离子浓度。在光催化合成开始之前,TiO2薄膜表面附近的Ag+离子浓度等于溶液中Ag+离子的平均浓度。因此,溶液浓度决定了光催化合成初期TiO2薄膜表面附近Ag原子的浓度。还原的Ag原子既可以吸附在薄膜表面,也可能脱附重新进入溶液。而吸附在TiO2薄膜表面上的Ag原子有可能被空穴重新氧化成Ag+离子而重新进入溶液。考虑到原子和离子扩散的影响,我们认为光催化初期的Ag原子浓度如图8(a)所示。一旦Ag原子在TiO2薄膜表面附近的局域浓度过饱和,成核过程开始。所以,AgNO3溶液浓度越高,晶核的尺寸越小,晶核密度越大是必然的。我们的结果也充分证明了这一结论。但现有的晶体生长理论无法解释晶核尺寸和密度不同所导致的生长模式的改变。

图8 光催化合成金属Ag纳米颗粒的生长机制示意图:(a)还原Ag原子的密度分布,(b)Ag初级晶核的密度分布,(c)Ag纳米片的密度分布Fig.8 Schematic illustration of photocatalytic growth of Ag nanostructures on titania films in AgNO3solution:(a)Photocatalytic reduction,(b)Nucleation of primary particles,(c)self-assembly growth via oriented attachment
我们认为,光催化合成金属Ag纳米颗粒过程中生长模式的改变主要与薄膜表面附近Ag原子的不均匀分布有关。当AgNO3溶液浓度较低时,由于Ag原子的产生率较小,所以薄膜表面的Ag原子的过饱和度相对较小,晶核尺寸较大,晶核密度较小,但分布较宽,因此晶核的长大更接近均匀分布的晶核长大过程,即OR生长机制。当AgNO3溶液浓度较高时,Ag原子的产生率较大,使得薄膜表面附近的Ag原子尚未充分扩散就已经达到了较高的过饱和度,因此晶核的分布沿薄膜法向方向是高度不均匀的,如图8(b)所示。由于平行于膜面方向的晶核密度较大,而尺寸差异较小,从而导致晶核优先沿平行于膜面方向发生联并。同时,由于此时的晶粒尺寸较小,晶核比较容易调整联并方向使得自由能最低,从而出现了OA机制主导的各向异性生长,如图8(c)所示。从图2的SEM照片中可以看出,金属Ag纳米片显然不是异质成核生长的结果,这有利支持了我们关于Ag纳米片形成的物理机制。OA生长机制可能不仅仅发生在晶核之间或晶核与Ag纳米片之间,而且也可能发生在Ag纳米片之间,这样才可能对金属Ag纳米片形貌的演化行为给出正确的解释。
3 结 论
本文用简单的光催化还原方法,通过控制AgNO3浓度,在TiO2薄膜表面分别得到了Ag的不规则的类球形和片状纳米颗粒。通过SEM和原位的消光光谱研究了Ag在TiO2薄膜表面的光催化生长行为。研究结果发现:光催化合成金属Ag纳米颗粒存在着两个完全不同的生长机制,即OR机制和OA机制,分别对应着金属Ag纳米颗粒的各向同性和各向异性生长。较高的AgNO3浓度更有利于OA机制的产生,从而使Ag纳米颗粒各向异性生长成为片状颗粒。在此基础上,本文提出了有关光催化生长金属Ag纳米颗粒的物理模型。这个模型表明:AgNO3溶液的浓度决定了Ag初级晶核的大小和局域密度,从而达到了对纳米颗粒不同的生长动力学行为的控制。
[1]Dieringer J A,Wustholz K L,Masiello D J,et al.J.Am.Chem.Soc.,2009,131(2):849-854
[2]Tam F,Goodrich G P,Johnson B R,et al.Nano Lett.,2007,7:496-501
[3]Liu M,Qu S W,Yu W W,et al.Appl.Phys.Lett.,2010,97(23):1906-1908
[4]Haruta M.Gold Bull.,2004,37(1-2):27-36
[5]Tripp R A,Dluhy R A,Zhao Y P.Nano Today,2008,3(3/4):31-37
[6]Wu H P,Liu J F,Wu X J,et al.Int.J.Adhes.Adhes.,2006,26:617-621
[7]TONG Jian-Bo(佟建波),HUANG Xi(黄茜),ZNANG Xiao-Dan(张晓丹),et al.Acta Phys.Sin.(物理学报),2012,61(4):2753-2759
[8]YU Mei(于美),LIU Peng-Rui(刘鹏瑞),SUN Yu-Jing(孙玉静),et al.J.Inorg.Mater.(无机材料化学),2012,27(1):89-94
[9]WANG En-Hua(王恩华),LIU Su-Wen(刘素文),LI Tang-Gang(李堂刚),et al.Chinese.J.Inorg.Chem.(无机化学学报),2011,27(3):537-541
[10]Lu Y,Liu G L,Lee L P.Nano Lett.,2005,5:5-9
[11]Zhang J T,Li X L,Sun X M,et al.J.Phys.Chem.B,2005,109(25):12544-12548
[12]Mock J J,Barbic M,Smith D R,et al.J.Chem.Phys.,2002,116:6755-6759
[13]Kelly K L,Coronado E,Zhao L L,et al.J.Phys.Chem.B,2003,107:668-677
[14]LAI Wen-Zhong(赖 文 忠),ZHAO Wei(赵 威),YANG Rong(杨容),et al.Acta Phys.-Chim.Sin.(物理化学学报),2010,26(4):1177-1183
[15]WANG Yue-Hui(王悦辉),ZHANG Qi(张琦),WANG Ting(王婷),et al.Chinese.J.Inorg.Chem(无机化学学报)2010,26(3):365-373
[16]Kulkarni A P,Noone K M,Munechika K,et al.Nano Lett.,2010,10(4):1501-1505
[17]Nishijima Y,Ueno K,Yokota Y,et al.J.Phys.Chem.Lett.,2010,1(13):2031-2036
[18]Christopher P,Ingram D B,Linic S.J.Phys.Chem.C,2010,114(19):9173-9177
[19]Sun Y,Mayers B,Herricks T,et al.Nano Lett.2003,3(7):955-960
[20]Lim B,Yu T,Park J,et al.Angew Chem.Int.Ed.,2011,123(27):6176-6179
[21]Liu Z,Zhou H,Lim Y S,et al.Langmuir,2012,28(25):9244
[22]Banfield J F,Welch S A,Zhang H Z,et al.Science,2000,28(9):751-754
[23]Li D S,Nielsen M H,Lee J R I,et al.Science,2012,336(6084):1014-1018
[24]Zhang Q,Liu S J,Yu S H,et al.J.Mater.Chem.,2009,19(2):191-207
[25]Liang H Y,Zhao H G,Rossouw D,et al.Chem.Mater.,2012,24(12):2339-2346
[26]Lu L H,Kobayashi A,Kikkawa Y,et al.J.Phys.Chem.B,2006,110(46):23234-23241
[27]Wang R,Liu D,Zuo Z,et al.J.Mater.Chem.,2012.22:2410-2418
[28]Mills A,Hill G,Stewart M.Appl.Spectrosc.,2004,58(8):922-928
[29]Tanabe I,Matsubara K,Standridge S D,et al.Chem.Commun.,2009,24:3621-3623
[30]Matsubara K,Kelly K L,Sakai N,et al.J.Mater.Chem.,2009,19:5526-5536
[31]Jin R,Cao Y C,Hao E,et al.Nature,2003,425(2):487-490
[32]Ung T,Liz-Marzan L M,Mulvaney P.J.Phys.Chem.B,2001,105(17):3441-3452
[33]Moores A,Goettmann F.New J.Chem.,2006,30:1121-1132
[34]Ohring M.Materials Science of Thin Films:Deposition and Structure.San Diego(USA):Academic Press,2002:45
猜你喜欢
中国氯碱(2023年1期)2023-02-22
化工学报(2022年7期)2022-08-10
合成材料老化与应用(2022年3期)2022-06-27
上海大学学报(自然科学版)(2020年2期)2020-05-13
陶瓷学报(2019年6期)2019-10-27
科技视界(2019年11期)2019-06-20
陶瓷学报(2019年5期)2019-01-12
北极光(2016年4期)2016-06-06
中国资源综合利用(2016年9期)2016-01-22
应用化工(2014年11期)2014-08-16
