
含联苯三羧酸及二咪唑基吡啶配体的两个锌配位聚合物的合成、结构及荧光性质
2014-01-02 00:54王淑菊田彦文由立新孙亚光
无机化学学报 2014年3期
王淑菊 田彦文 由立新 丁 茯 孙亚光*,
(1沈阳化工大学应用化学学院,沈阳 110142)
(2东北大学材料与冶金学院,沈阳 110004)
0 引 言
配位聚合物(Coordination Polymers)通常是由金属离子与含氧、含氮等多齿有机配体通过自组装过程形成的具有一维、二维或三维,并且有重复性网状结构的晶体材料[1-3]。因为配位聚合物具有高比表面积、高孔隙率、及其化学稳定性和热稳定性,所以它们在气体储存、吸附和分离,发光,磁性,催化和药物缓释等方面显示出巨大的潜在应用价值,引起了研究人员的广泛关注[4-10]。在配位聚合物的构筑过程中,有机配体在金属中心之间起着桥联作用。特别是芳香羧酸因其在结构上具有一定的刚性和稳定性,是构筑配位聚合物常用配体[11-12]。联苯羧酸类配体具有苯环的刚性骨架结构,而且两苯环间C-C键可以旋转,从而表现出一定的柔性,可从不同方向与金属中心形成空间结构。同时,羧基中的氧原子易于形成氢键,有助于配位聚合物构筑新型多维结构[13]。以对称联苯羧酸为配体构筑配位聚合物的相关报道较多[14-15],而非对称联苯羧酸配体报道相对较少。由于羧基的不对称,可能形成更为新颖的空间结构[16]。
另外,加入辅助配体可以更好地协同主配体对目标产物进行调控[17]。2,6-二(1-咪唑基)吡啶是具有三个杂环的半刚性配体,分子内的两个C-N键可以旋转,形成折叠或螺旋结构[18],与芳香羧酸配合使用能够灵活地构筑不同结构的配位聚合物[19]。
本文采用 3,3′,5-联苯三羧酸(H3bpta)和 2,6-二(1-咪唑基)吡啶(bip)为配体与锌盐反应,在水热条件下合成了两个配位聚合物{[Zn3(H2O)7(bpta)2]·5H2O}n(1)和{[Zn2Cl(bpta)(bip)2]·2H2O}n(2)。单晶 X-射线结构分析表明,化合物1形成1D链状结构,化合物2在辅助配体的调控下网联成2D网状结构,并进一步研究了1和2的热稳定性和荧光性质。
1 实验部分
1.1 试剂与仪器
所有试剂均为分析纯试剂,未经进一步纯化处理。元素分析使用Perkin-Elemer 470型元素分析仪;红外光谱使用Nicolet IR-700型红外光谱仪,波长 4 000~400 cm-1,KBr压片; 热重分析 (TGA)在NETZSCH TG 209热重分析仪上氮气中进行测定;Perkin-Elmer LS55型荧光分光光度计进行荧光分析;单晶测定是采用Bruker SMART 1000 CCD面探衍射仪。
1.2 配位聚合物的合成
1.2.1 {[Zn3(H2O)7(bpta)2]·5H2O}n(1)的合成
H3bpta(0.105 mmol)、Zn(NO3)2·6H2O(0.3 mmol)、KOH(0.3 mmol)、4 mL乙醇和3 mL H2O混合均匀,置于密封的25 mL聚四氟乙烯内衬不锈钢反应釜中,在90℃下反应72 h,5℃·h-1的速率降至室温,得到无色透明长条型晶体,用蒸馏水洗净,在空气中晾干。晶体不溶于水和一般的有机溶剂。产率为46.80%(以 Zn(NO3)2·6H2O 计), 化学式 C30H34O24Zn3(Mr=974.68)。 元素分析测定值(%):C,36.37;H,3.29。理论值(%):C,36.96;H,3.52。 IR 主要吸收峰(cm-1)为:3 404b、1 615m、1 568s、1 422m、1 382m、1 277w、1 072w、834m、764s、728m、690m、631w、588w。
1.2.2 化合物{[Zn2Cl(bpta)(bip)2]·2H2O}n(2)的合成
将 ZnCl2(0.6 mmol)、H3bpta(0.2 mmol)、bip(0.2 mmol)、8 mL H2O和2 mL DMF混合均匀,置于密封的25 mL聚四氟乙烯内衬不锈钢反应釜中,在150℃下反应72 h,5℃·h-1的速率降至室温,得到淡黄色长条型晶体,用蒸馏水洗净,在空气中晾干。晶体不溶于水和一般有机溶剂。产率为51.25%(以ZnCl2计),化学式C37H27ClN10O7Zn2(Mr=889.88)。元素分析实验值(%):C,49.56;H,3.55;N,15.08;理论值(%):C,49.94;H,3.05;N,15.74。 IR 主要吸收峰(cm-1)为:3 422b、1 608s、1 502s、1 466m、1 350m、1 182m、1 087 m、1 013m、943m、850m、800m、765s、693w、647w。
1.3 晶体结构测定
化合物晶体结构测定在293 K下采用Bruker SMART 1000型X-射线衍射仪,石墨单色器单色化Mo Kα 射线(λ=0.710 73 nm)作入射光,以 ω-2θ扫描方式收集衍射点。晶体结构均采用直接法解析,最小二乘法F2精修,使用SHELXS-97和SHELXL-97程序包[20-21]。非氢原子坐标用直接法解出,并对他们的坐标及其各向异性热参数进行全矩阵最小二乘法修正。氢原子的位置通过理论加氢得到,并使用固定的各向异性热参数加入结构精修。在化合物1中,水分子O8处于二重轴位置,所以其占有率为0.5; 水 分 子 O7,O11,O12,O13,O14,O15,O16 和O17无序,所以精修时利用了裂分的方法并采用ISOR和EADP等指令。化合物晶体学数据在表1列出。
CCDC:929651,1;CCDC:929652,2。

表1 化合物1和2的主要晶体学数据Table 1 Crystal data and structure refinement for complexes 1 and 2
2 结果与讨论
2.1 配位聚合物晶体结构
2.1.1 化合物{[Zn3(H2O)7(bpta)2]·5H2O}n(1)晶体结构描述
单晶结构分析表明,化合物1的不对称单元中包含1.5个晶体学独立的Zn(Ⅱ)离子,1个bpta配体分子,3.5个配位水分子和2.5个游离水分子。结构中Zn1分别与两分子bpta配体中3-COO-的O4和5-COO-的O5a及2个水分子O9、O10配位形成四配位三角锥构型,Zn1位于三角锥的几何中心。O4位于三角锥的轴向顶点,O9、O5a和O10在同一平面。Zn2位于二重轴特殊位置,与bpta配体3′-COO-的 O1,O1b 及 3 个水分子中 O7/O11、O7b/O11b、O8形成五配位三角双锥构型。其中O1、O1b、O8/O8b位于赤道面,O7/O11和O7b/O11b位于轴心。bpta配体的 3 个羧基均 以 μ1-η1∶η0方式与 Zn 配位(scheme 1),2个Zn1通过两分子bpta配体3-COO-,3′-COO-连接形成环状结构,再以Zn2为节点通过bpta配体5-COO-连接,沿a轴方向延伸,形成1D无限链(图2)。在晶体结构中,bpta配体的羧基与配位水分子以O-H…O氢键作用使化合物形成3D超分子结构(表 2)。

Scheme 1 Binding mode of bpta ligand
2.1.2 化合物{[Zn2Cl(bpta)(bip)2]·2H2O}n(2)晶体结构描述

图1 化合物1中Zn(Ⅱ)配位环境Fig.1 Coordination environment of Zn(Ⅱ)in complex 1
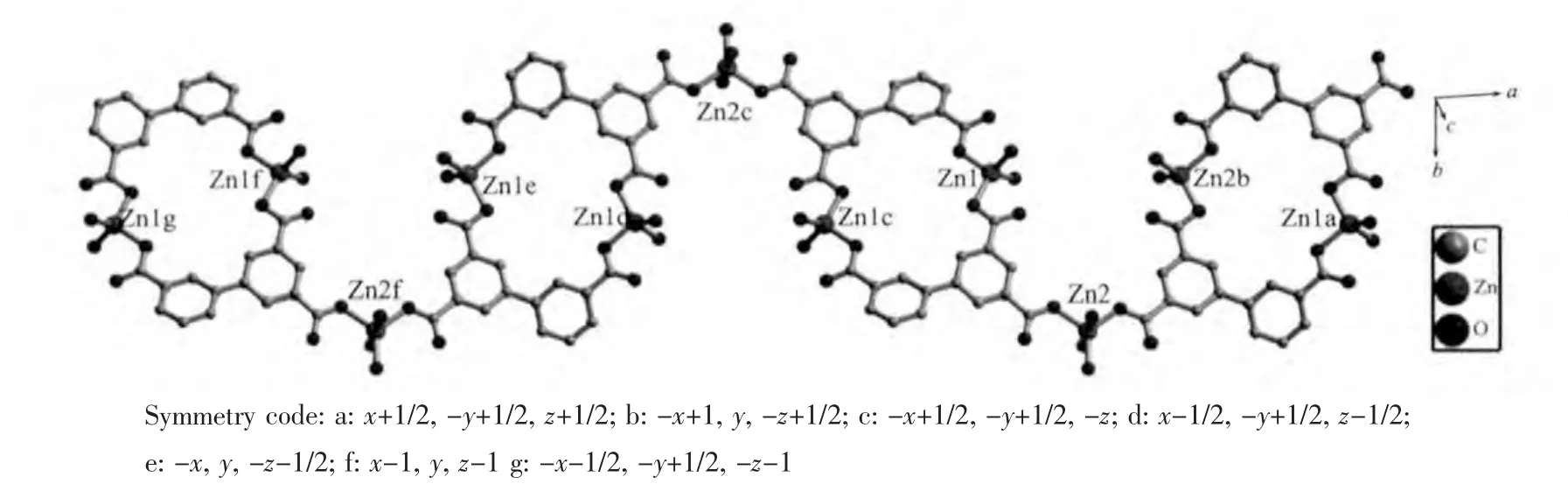
图2 化合物1的1D链状结构Fig.2 Views of 1D chain structure of the complex 1

表2 化合物1的氢键键长和键角Table 2 Seletrd hydrogen bonds for complex 1
如图3所示,化合物2结构基元中包含3个晶体学独立的Zn(Ⅱ)离子(其中Zn2和Zn3位于二重轴的位置上),1个bpta配体分子,2个bip配体分子及1个配位Cl原子。结构中Zn(Ⅱ)离子是四配位的,Zn1与2个bip配体的N1、N6原子及 2个bpta配体3′-COO-的O1和5-COO-的O3d原子配位, 形成一个扭曲的三角锥。N1位于三角锥的轴向顶点,N6、O1和O3d原子位于底面位置。Zn2与2个bip配体的 N3、N3b原子及 2个 bpta配体 3-COO-的O5c、O5d原子配位,形成扭曲的三角锥。配体bpta的羧基以 μ1-η1∶η0方式与 Zn(Ⅱ)离子配位。 Zn3 与Zn1和Zn2相似,处于一个扭曲的三角锥的中心,4个配位原子分别来自于2个bip配体的N9、N9a及2个配位Cl1、Cl1a。相邻的Zn(Ⅱ)通过bip配体中咪唑基的N原子桥连形成1D螺旋链状结构 (如图3所示)。bpta配体3′-COO-的O原子与相邻1D链中Zn(Ⅱ)配位,在ac面上形成2D网状结构。由于螺旋链间的相互作用,化合物2没有扩展成3D框架(如图4所示)。

图3 化合物2中Zn(Ⅱ)配位环境Fig.3 Coordination environment of Zn(Ⅱ))in complex 2
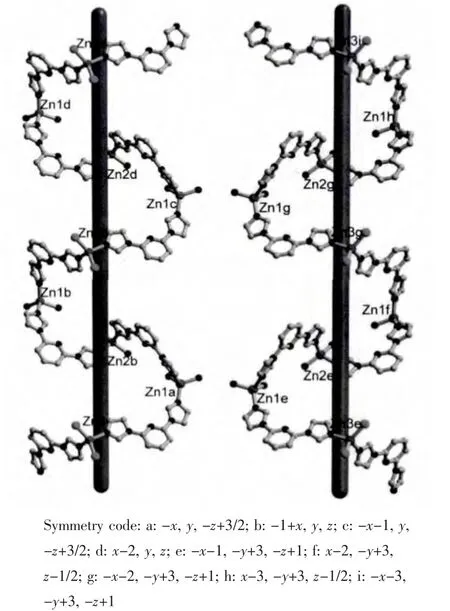
图4 化合物2的1D链状结构Fig.4 Views of 1D chain structure of the complex 2
2.2 热稳定性分析
热重分析(TGA)采用NETZSCH TG 209热重分析仪,在氮气保护中进行测定,升温速率为10℃·min-1,温度范围 25~1 000 ℃。化合物 1在 25~160 ℃范围发生第一次失重,约占总重的14.07%,为化合物1中游离水和部分配位水分子。随着温度升高,配位水分子继续失去,呈现出160~350℃的失重范围,约占总重的7.81%,至此全部失重约为21.88%,与结构中游离水分子和配位水分子所占的比重之和(理论值为22.15%)相近。在350~830℃附近是第三次失重范围,配体bpta逐渐分解,骨架坍塌。最后恒定在25.15%,残余物为ZnO(理论值为25.04%)。化合物2在25~135℃附近是第一次失重范围,占总重的4.70%,与化合物2中2个游离水分子(理论值为4.05%)相近。在135~350℃范围内化合物2没有发生失重现象,说明其框架结构能保持稳定。从350~920℃是第二次失重范围,骨架迅速坍塌,配体bpta和bip逐渐分解。最后恒定在18.32%,残余物为ZnO(理论值为18.29%)。热重分析结果表明,化合物2的二维框架结构的稳定温度可达到350℃,具有较高的热稳定性。
2.3 红外光谱分析
化合物1在3 403 cm-1处的宽峰为O-H伸缩振动峰,而且由于氢键的缔合作用使峰变宽。配体H3bpta在 1704、1626、1427 和 1282 cm-1处的吸收峰, 在化合物 1中位移至 1 615、1 568、1 422和1 382 cm-1处,说明羧基氧与Zn配位。化合物2在3 422 cm-1处的宽峰为O-H伸缩振动峰,配体H3bpta于1704cm-1处的特征吸收峰νC=O(COOH)在形成配位聚合物后消失,出现了羧酸根(-COO-)的反对称νas(COO-)1 502 cm-1和对称伸缩振动吸收峰νs(COO-)1 466 cm-1,表明羧基中的氧原子参与了配位。在1 608 cm-1吸收峰为咪唑C=N伸缩振动吸收峰。
2.4 荧光光谱分析
室温下,配体H3bpta、bip和化合物1和2的固态荧光光谱如图5所示。当在330 nm波长的光源下激发时,H3bpta配体在440 nm发射荧光;在激发波长为370 nm时,bip配体得到453 nm的发射荧光;当在330 nm波长的光源下激发时,化合物1在440 nm处出现荧光发射峰;当在370 nm波长的光源下激发时,化合物2在450 nm处出现荧光发射峰。荧光强度比较:bip>化合物2>H3bpta>化合物1。

图5 室温下化合物1和2的固态荧光光谱Fig.5 Solod-state fluoresecent emission spectrum of complexes 1 and 2 at room temperature
配体H3bpta和bip都具有较大的共轭结构,都可以吸收激发能,并产生荧光发射。化合物1的荧光强度低于H3bpta,这是因为配体与Zn(Ⅱ)的键合,促使配体分子内C-C键扭转,柔性表现增强,内部电荷转移增加,能量损失增多[22];而且分子内多个配位水分子的存在,以热振动的形式损失能量,使得荧光辐射能量减弱。化合物2的荧光强度高于配体H3bpta和化合物1,是因为bip配位到Zn(Ⅱ)上,阻止了溶剂水分子的配位,降低了水分子以热振动的形式损失能量,使其有较好的发光性质[23];但是,C-N键发生扭转,配体bip柔性增强而损失部分能量,使化合物2荧光强度低于配体bip。
[1]Antek G,Foy W,Yaghi O M,et al.J.Am.Chem.Soc.,2006,128(11):3494-3495
[2]Plater M J,Foreman M R,Gelbrich T,et al.Dalton Trans.,2000:1995-2000
[3]Düren T,Sarkisov L,Yaghi O M,et al.Langmuir,2004,20(7):2683-2689
[4]Murray L J,Dinc M.Chem.Soc.Rev.,2009,38(5):1294-1314
[5]Li J R.,Kuppler R.J.Chem.Soc.Rev.,2009,38(5):1477-1504
[6]Allendorf M D,Bauer C A.Chem.Soc.Rev.,2009,38(5):1330-1352
[7]Rosseinsky M.J.Microporous Mesoporous Mater.,2004,73(1-2):15-30
[8]Kurmoo M.Chem.Soc.Rev.,2009,38(5):1353-1379
[9]Meek S T,Greathouse J A.Adv.Mater.,2011,23(2):249-267
[10]Lee J Y,Farha O K.Chem.Soc.Rev.,2009,38(5):1450-1459
[11]Tian D,Pang Y,Zhou Y.H,et al.CrystEngComm,2011,13(3):957
[12]JIN Jing(金晶),LIU Yong-Hua(刘永华),NIU Shu-Yun(牛淑云),et al.Chinese J.Inorg.Chem.(无机化学学报),2013,29(3):455-464
[13]Rowsell J L C,Yaghi O M,Chen B L,et al.Angew.Chem.Int.Ed.,2005,44(30):4647-4649
[14]Guo Z Y,Xu H,Su S Q,et al.Chem.Comm.,2011,47:5551-5553
[15]Li L N,Zhang S Q,Han L,et al.Crystal.Growth Des.,2013,13(1):106-110
[16]Chang X H,Qin J H,Lu-Fang,et al.Crystal.Growth Des.,2012,12(9):4649-4657
[17]Xu J,Yao X Q,Huang L F,et al.CrystEngComm,2011,13(3):857-865
[18]Chen C Y,Cheng P Y,Wu H H,et al.Inorg.Chem.,2007,46:5691-5699
[19]Lee J Y,Chen C Y,Lee H M,et.al.Crystal.Growth Des.,2011,11(4):1230-1237
[20]Sheldrick G M.SHELXS-97,Program for X-ray Crystal Structure Solution,Göttingen University,Germany,1997.
[21]Sheldrick G M.SHELXS-97,Program for X-ray Crystal Structure Refinement,Göttingen University,Germany,1997.
[22]GUO Yin-Chen(郭应臣),CHEN Shu-Yang(陈书阳),QIU Dong-Fang(邱东方),et al.Chinese J.Inorg.Chem.(无机化学学报),2011,27(8):1517-1522
[23]LI Gong-Chun(李公春),WANG Hong-Sheng(王宏胜),ZHU Lei(朱蕾),et al.Chinese J.Inorg.Chem.(无机化学学报),2012,28(11):2369-2372
猜你喜欢
科技创新与应用(2022年4期)2022-03-04
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21
教育与职业(下)(2019年7期)2019-08-15
中国管理信息化(2018年7期)2018-05-27
衡阳师范学院学报(2016年3期)2016-07-10
山东工业技术(2016年7期)2016-04-08
应用化工(2014年10期)2014-08-16
应用化工(2014年7期)2014-08-09
火炸药学报(2014年3期)2014-03-20
