
家族性局灶节段肾小球硬化症还是Alport综合征?
2013-12-25 08:45:43刘小荣
中国循证儿科杂志 2013年6期
凌 晨 刘小荣 陈 植 沈 颖
·病例讨论·
家族性局灶节段肾小球硬化症还是Alport综合征?
凌 晨 刘小荣 陈 植 沈 颖
1 病例资料
患儿,男,12.5岁,因“间断双下肢水肿4月余”于2013年5月25日入首都医科大学附属北京儿童医院(我院)。
入我院前4个月余,患儿家长无意中发现患儿眼睑、腿部水肿。患儿无发热、咳嗽和皮疹等不适。就诊于当地医院,尿常规:蛋白+++,潜血微量。血生化示白蛋白:25.2 g·L-1,胆固醇9.83 mmol·L-1,BUN 3.25 mmol·L-1,SCr 50.5 μmol·L-1,诊断为肾病综合征。予甲泼尼龙1.6 mg·kg-1,每日1次口服,期间多次复查尿蛋白均++~+++,潜血在-~+。激素治疗41 d自行减停。入我院前10 d曾就诊于另一医院,尿常规:蛋白+++,潜血++,24 h尿蛋白定量8.32 g,血生化:白蛋白17.6 g·L-1、胆固醇8.18 mmol·L-1、BUN 5.8 mmol·L-1和SCr 44.7 μmol·L-1。甲泼尼龙加量至48 mg,每日1次。尿蛋白未减轻。
患儿系G1P1。母孕期体健,否认放射性物质及化学毒物接触史,否认家族近亲结婚史。患儿既往体健,智力及体格发育同正常同龄儿。
入院查体:神清,精神可。发育正常,面色红润,全身未见皮疹及出血点,卡疤阳性,浅表淋巴结未触及肿大,双眼睑及颜面未见水肿,咽无充血,双侧扁桃体无肿大,双肺呼吸音粗,未闻及干湿性啰音,心音有力,律齐,各瓣膜区听诊未闻及杂音。腹软,无压痛反跳痛及肌紧张,肝、脾肋下未扪及,双肾区无叩痛,肠鸣音正常,神经系统查体未见异常,双下肢未见水肿。
辅助检查:血常规:WBC 10.94×109·L-1,RBC 3.87×1012·L-1,N 0.36,L 0.68,Hb 113 g·L-1,PLT 334×109·L-1,CRP<8 mg·L-1。尿蛋白/尿肌酐2.08。血生化:总蛋白37.9 g·L-1、白蛋白23.7 g·L-1、球蛋白14.2 g·L-1、BUN 5.10 mmol·L-1、SCr 61.30 μmol·L-1、总胆固醇8.56 mmol·L-1和三酰甘油4.42 mmol·L-1。凝血功能筛查正常。ESR 11 mm·h-1。24 h尿蛋白定量9.1 g。ANCA、ds-DNA和ANAs均阴性。乙肝、丙肝、梅毒和HIV均阴性。CD系列:NK-C 1.4%。听力及视力检查均未见异常。血氨基酸分析和尿代谢筛查均未见异常。
肾活检光镜报告:可见10个肾小球,1个球性硬化,其余肾小球系膜细胞和基质轻度弥漫增生,其中3个阶段性硬化,足细胞增生;肾小管上皮中度空泡变性,灶状萎缩,肾间质灶状淋巴细胞和单核细胞浸润伴纤维化;小动脉管壁增厚(图1A)。电镜报告:肾小球系膜区无增生扩大,毛细血管壁基底膜无增厚或变薄,未见分层或疏松区,未见电子致密物沉积,足突细胞节段性足突融合(图1B , C) 。 免疫荧光皮肤基底膜Ⅳ型胶原α链检测未见异常。符合非特殊型局灶节段肾小球硬化症(FSGS)。
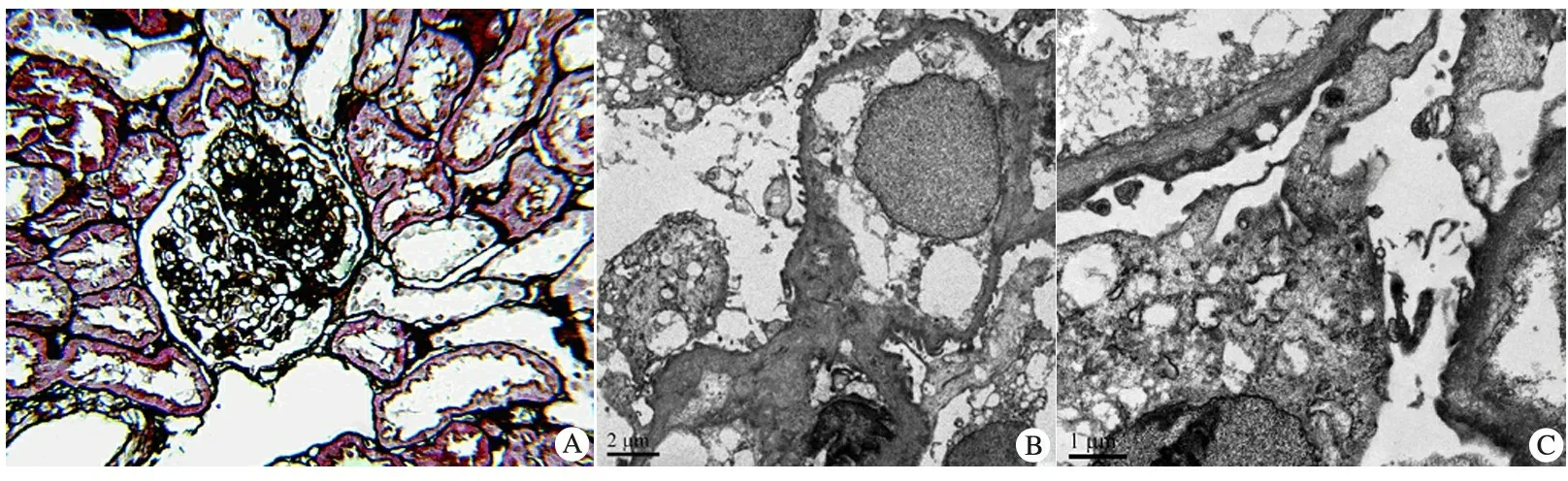
图1 患儿肾组织病理学检查所见
Notes A: 光镜下见10个肾小球,1个球性硬化,其余肾小球系膜细胞和基质轻度弥漫增生,其中3个阶段性硬化,足细胞增生;肾小管上皮中度空泡变性,灶状萎缩,肾间质灶状淋巴细胞和单核细胞浸润伴纤维化;小动脉管壁增厚(PASM,×200);B(×6 700),C(×8 000):电镜下见肾小球系膜区无增生扩大,毛细血管壁基底膜无增厚或变薄,未见分层或疏松区,未见电子致密物沉积,足突细胞节段性足突融合
予他克莫司1.5 mg,甲泼尼龙48 mg·d-1,每日2次口服,治疗半月余,复查24 h尿蛋白定量3.1 g。随访9个月,患儿尿常规蛋白波动于+~+++。肾功能未见异常。
询问患儿家族史,发现患儿家系中有多名类似症状的患者,图2患儿家系图谱显示,本患儿为该家系中先证者(Ⅳ1)。Ⅲ1 10岁时出现水肿,无肉眼血尿,尿常规蛋白+++,潜血阴性。诊断肾病综合征,予抗炎消肿治疗后康复,目前尿常规及肾功能均未见异常。Ⅱ1 30岁时出现眼睑及双下肢水肿,无肉眼血尿,诊断“肾病”,予抗炎治疗后水肿消退,未再出现类似症状,目前60岁,尿常规正常,血生化未见肾脏功能降低。Ⅲ5 10岁时出现水肿及尿检异常,13岁诊断“尿毒症”死亡。Ⅳ9 11岁时出现眼睑及双下肢水肿,尿常规蛋白++++,潜血微量,24 h尿蛋白提示大量蛋白尿,诊断肾病综合征,应用激素规律治疗1年余,近期24 h尿蛋白仍为大量蛋白尿,肾活检病理诊断为FSGS(非特殊型)。Ⅳ10 5岁,目前体健。Ⅲ19症状同Ⅲ5,13岁发病,16岁时因尿毒症死亡。Ⅱ5 26岁时出现水肿症状,未予规律治疗,30岁时因尿毒症死亡。Ⅱ3、Ⅱ8、Ⅱ10及Ⅲ15为该家系中身体健康的女性,但其子辈或孙辈中有该病患者。该家系中无任何一名成员出现肉眼血尿,视力异常、听力障碍和肢体异常等其他伴随症状。

图2 患儿家系图谱
2 讨论
FSGS多通过肾活检诊断。如该患儿无异常家族史,那么诊断FSGS应无异议。但鉴于患儿阳性家族史,有明显的遗传倾向(系谱分析X连锁可能性大),是否能够除外Alport综合征。
FSGS是导致终末期肾衰竭的主要肾小球疾病。家族性FSGS 是FSGS 中一种特殊类型,表现为家族中多个成员均有发病。Daskalakis等[1]研究发现超过18%的FSGS 有家族聚集现象。对于FSGS的发病机制较明确的是,编码足细胞裂孔膜蛋白nephrin、podocin和细胞骨架蛋白a-actinin4等的基因突变是家族性和散发性肾病的重要原因[2~5]。
本例家族中受累成员均有一过性水肿表现,先证者临床表现为大量蛋白尿、低白蛋白血症、高脂血症及水肿,符合肾病综合征(肾炎型)表现,先证者和Ⅳ9肾脏病理均为FSGS(非特殊型)。但需要除外其他疾病导致的继发性FSGS。患儿家系分析显示伴X连锁可能性大。需要注意的是文献报道的家族性FSGS,遗传方式均为常染色体显性遗传和常染色体隐性遗传,而不伴性X染色体遗传[6]。FSGS伴X染色体的遗传方式文献未见报道。
Alport综合征是以血尿、感音神经性耳聋和进行性肾功能减退为特点的遗传性疾病。分为X连锁显性遗传型、常染色体隐性遗传型及常染色体显性遗传型。且X连锁型Alport综合征男性可出现蛋白尿,肾病综合征的发生率为30%~40%,北京大学第一医院报道蛋白尿达肾病综合征者占31.8%[7]。男性患者肾脏预后极差,几乎全部将发展为终末期肾病。
该家系多个女性携带者均有一过性临床症状(Ⅱ1,Ⅲ1),男性患者预后明显差于女性患者,符合X连锁显性遗传的Alport综合征表现。但本例家系中各受累成员均以水肿、蛋白尿为主要表现,而无听力障碍及眼部病变等其他表现。部分女性携带者(Ⅱ3、Ⅱ8、Ⅱ10、Ⅲ15)无任何临床症状。皮肤活检Ⅳ胶原α链免疫荧光检查及肾活检电镜下均未见典型Alport综合征表现。需要指出的是Alport综合征不同家系及受累者表现存在差异。肾活检电镜诊断目前仍具有十分重要的地位。但部分高度怀疑为Alport综合征的患者,可能肾小球基底膜无典型的病理改变,另外有些家系临床表现为进行性肾功能减退,有COL4A5基因突变,但肾小球基底膜仅有变薄表现。故肾小球基底膜无典型表现,不能作为Alport综合征的除外证据。由于某些确诊的X连锁型Alport综合征患者或基因携带者,可有基底膜α5(Ⅳ)链的正常表达,因而皮肤活检正常并不能除外Alport综合征的诊断[8]。故该病究竟是Alport综合征还是家族性FSGS新的遗传方式,有待于进一步基因分析。
本文病例值得总结经验如下:①不应仅关注肾病综合征的患儿是否存在阳性家族史,更应该关注阳性家族史的细节部分,如遗传方式,各受累成员的表现、诊断、治疗及预后等内容。这些信息对诊断及治疗具有重要的指导价值。②诊断FSGS的同时必须除外继发性病因导致FSGS发病的可能。③由于Alport综合征是一种进行性损伤性疾病,对于家系中的患者需注意随访观察,如随访过程中部分成员出现典型临床表现,对于明确诊断具有一定的价值。④对于家族中有以肾脏病家族史的患儿检查不能仅限于肾活检结果,基因检测对于明确诊断及产前指导具有重要的价值[9]。
对于该家系应进行长期追踪,观察家系中患者的疾病进展及其他正常家族人员的健康情况,针对可疑染色体进行有针对性的基因组扫描,若能发现可疑致病基因,则有助于进一步对该病明确诊断。
[1]Daskalakis N, Winn MP.Focal and segmental glomerulosclerosis.Cell Mol Life Sci,2006,63(21):2506-2511
[2]Kaplan J, Pollak MR.Familial focal segmental glomerulosclerosis.Curr Opin Nephrol Hypertens,2001,10(2):183-187
[3]Kaplan JM, Kim SH, North KN, et alMutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis.Nat Genet,2000,24(3):251-256
[4]Caridi G, Bertelli R, Di Duca M, et al. Broadening the spectrum of diseases related to podocin mutations.J Am Soc Nephrol,2003,14(5):1278-1286
[5]Dai SC(戴胜川), Wang CH, Pan XX, et al. ACTN4 gene variation and polymorphism in patients with primary focal segmental glomerulosclerosis. Chin J Nephrol(中华肾脏病杂志),2008,24(2):108-114
[6]Conlon PJ,Lynn K, Winn MP, et al. Spectrum of disease in familial focal and segmental glomerulosclerosis. Kidney Int, 1999, 56(5):1863-1871
[7]Wang F(王芳), Ding J, Yu LX, et al.Clinical characteristic patterns of patients with Alport syndrome in China.J Clin Pediatr(临床儿科杂志), 2003,21(10):601-604
[8]Wang F, Zhao D, Jie D, et al. Skin biopsy is a practical approach for the clinical diagnosis and molecular genetic analysis of X-linked Alport's syndrome.J Mol Diagn, 2012, 14(6):586-593
[9]Chatterjee R,Hoffman M, Cliften P, et al. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of alport syndrome or proteinuria. PLos One,2013,10;8(10):e76360
10.3969/j.issn.1673-5501.2013.06.014
北京市科委首都临床特色应用研究-课题项目:Z121107001012052
首都医科大学附属北京儿童医院肾脏中心 北京,100045
刘小荣,E-mail:design2000@sina.com
2013-08-17
2013-11-27)
丁俊杰)
猜你喜欢
听力学及言语疾病杂志(2022年5期)2022-09-20 09:07:10
世界最新医学信息文摘(2021年12期)2021-06-09 08:37:12
振动与冲击(2018年4期)2018-03-05 00:34:24
临床医药文献杂志(电子版)(2017年71期)2017-03-10 15:35:28
实用临床医药杂志(2016年21期)2016-12-09 03:28:12
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
中国卫生标准管理(2015年1期)2015-01-26 21:17:41
