
固相萃取-气相色谱法测定人血中丙戊酸浓度的不确定度评定
2013-12-23 04:50彭静宋新文汪洋张华年徐华刘炘武汉市儿童医院湖北武汉430016
中国医院药学杂志 2013年20期
彭静,宋新文,汪洋,张华年,徐华,刘炘 (武汉市儿童医院,湖北 武汉430016)
丙戊酸是儿科最常用的抗癫痫药物之一,其体内代谢过程和疗效存在较大个体差异,为临床公认的需要进行血药浓度监测的品种,有效血药浓度范围为50~100 mg·L-1。参考已发表的研究报告[1-2],本实验建立了固相萃取-气相色谱(SPE-GC)法测定人血清中丙戊酸浓度的方法。该方法较其他方法能提高工作效率、减少试剂污染,可广泛推广。
我院开展丙戊酸血药浓度监测已有近10年历史,国内有关丙戊酸血药浓度监测的报道较多[3-5],但未见对测定方法不确定度评定的文献,这可能是与生物样品中药物浓度的检测步骤复杂、干扰因素较多有关。本实验以国家计量技术规范、不确定度评定的相关教材及文献为理论和方法指导[6,8-10],重点参考已发表文献[11-12]的评定步骤和内容,探讨了SPE-GC法测定人血清中丙戊酸浓度测定的不确定度评定方法。对主要影响因素进行分析评定,最终得出检测结果扩展不确定度值。可用于对丙戊酸血药浓度监测方法及测定结果准确度的评价。为更好控制血药浓度测定过程中关键步骤,开展癫痫患儿血药浓度监测提供有力依据。
1 材料
1.1 仪器 GC7890п型气相色谱仪(上海天美科学仪器有限公司);N-2000双通道色谱数据工作站,(浙江大学智能信息工程研究所);AB204电子分析天平(瑞士Mettler);LDZA-0.8A 低速自动平衡台式离心机(北京医用离心机厂)。ODS-SPE 柱,规格:200 mg/支(Agela Technologies公司)。
1.2 试药 丙戊酸对照品(美国Simga公司,CAS号99-66-1);正常空白人血清(实验室自制);甲醇为色谱纯试剂;水为超纯水。
2 方法
2.1 色 谱 条 件[2]ATSE-54 石 英 毛 细 管 色 谱 柱(21 m×0.53 mm,1.0μm),程序升温:初始温度60℃,维持时间5 min,升温速度10 ℃·min-1,最终温度180 ℃,维持时间5 min;进样口温度130 ℃;检测器FID,温度100 ℃;进样量3μL。
2.2 对照品溶液制备 精密称取丙戊酸对照品29.6 mg,置10 mL 量瓶中,加甲醇,配成贮备液。用5 mL单标线吸量管精密量取上述贮备液于10 mL量瓶中,以甲醇稀释成2 960,1 480,740,370,185μg·mL-1丙戊酸的系列标准溶液。
2.3 血清样品预处理 采用Agela ODS-SPE固相萃取小柱对血清样品进行处理。(1)活化:用2 mL甲醇润湿小柱;(2)水洗:用2 mL 超纯水淋洗小柱;(3)加样:在含药血清中加入0.01 mL 硫酸(6 mol·mL-1),用吸量管吸取0.5 mL 含药血清上样,然后用1 mL超纯水淋洗小柱去除样本中的内源性杂质和其他相关杂质,弃去废液。(4)收集洗脱液:用吸量管吸取0.5 mL 甲醇洗脱被分析物,收集洗脱液进样检测。
2.4 样品检测与计算 分别量取“2.2”项下丙戊酸的系列标准溶液各0.1 mL,加入到空白血清1 mL中混匀,制成系列含药标准血清(质量浓度分别为16.8,33.6,67.3,134.5,269.1μg·L-1),按“2.1”项下条件进行样品的测定,以丙戊酸信号的峰面积为纵坐标(A),含药标准血清浓度为横坐标(C,μg·L-1),得直线回归方程:A=aC +b。待测样品用同样的方法分析后,将样品信号的峰面积代入标准曲线,计算得到待测样品的浓度:C=,其中a为斜率,b为截距。
3 结果
3.1 不确定度来源分析 从检测过程和数学模型分析测定丙戊酸人血中含量的不确定度来源,主要有以下几个方面:(1)涉及对照品纯度和称重;(2)标准溶液的配制:涉及逐级稀释过程(影响因素包括吸量管、量瓶允差)的影响和温度等。(3)含药血清的配制:涉及量取标准溶液及空白血清的吸量管允差和温度的影响;(4)血清药物的提取:涉及回收率问题;(5)标准曲线的拟合;(6)仪器测定:涉及重复性问题。
3.2 不确定度分量的量化
3.2.1 A 类不确定度评定 ur(1)由仪器重复测定(精密度)引入的不确定度属于A 类不确定度。平行配制低(Low,L)和高(High,H)浓度质控样品各5份(n=5),按“2.3”项处理后进样分析,记录峰面积,按上述回归方程计算血药浓度值,为随机测定,属A 类不确定度。结果见表1。=16.8μg·mL-1=134.5μg·mL-1。
单次测量的实验标准差为:

观测列算术平均值的实验标准差为:
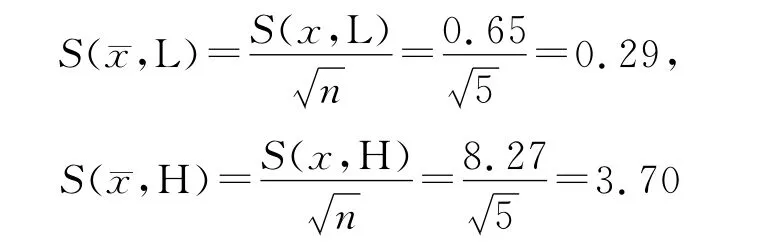
测定结果的标准不确定度为:
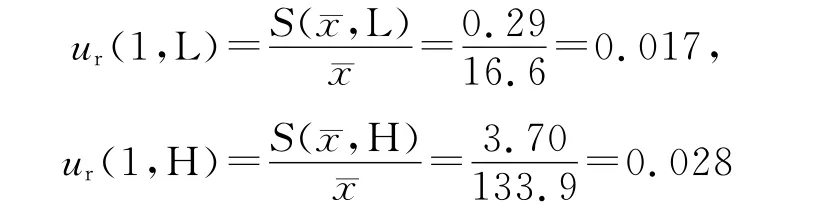
3.2.2 B类不确定度评定
(1)对照品称量引入的不确定度,ur(2) 对照品的相应质量由已扣除皮重的称量给出,则称量的3个不确定来源[10]:重复性、可读性(数字分辨率)以及由于天平校准产生的不确定度分量。其中天平校准本身有两个不确定度来源:灵敏度和校准函数的线性。灵敏度可忽略,因为称量是用同一架天平在很窄范围内进行。天平称量重复性引入的不确定度ur(W1),所用电子天平(d=0.01 mg)的检定证书给出的重复性误差为±0.2 mg,按矩形分布,则:ur(W1)=0.2/=0.12 mg
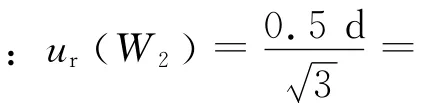
天平的线性引入的不确定度ur(W3),所用电子天平(d=0.01 mg)的检定证书给出的最大允差为±0.1 mg,按矩形分布,则:ur(W3)=0.1/0.058 mg
单次称量的标准不确定度:
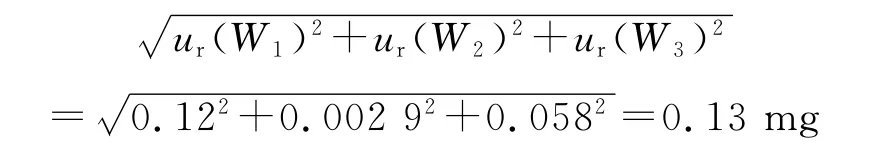
因称量过程采用减重法分两步,必须计算2次。
因而相对标准不确定度:

(2)配制对照品溶液时引入的不确定度,ur(3) 所用的10 mL量瓶中的溶液体积的不确定度主要来自:确定量瓶内部体积时的不确定度(校准);量瓶和溶液温度与量瓶体积校准时的温度不同(温度)。
校准:JJG196-2006规定,20 ℃时容量允差为±0.02 mL,按三角分布换算成标准偏差为=0.008 2 mL。
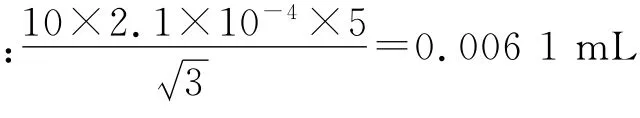
所用的5 mL 单标线吸量管体积的不确定度,与量瓶同理,包括:
校准:JJG196-2006 规定,20 ℃时的容量允差为±0.015 mL。按三角分布换算成标准偏差为0.015/=0.006 1 mL。
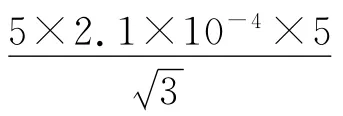
不考虑重复性误差,考虑到操作次数时,配制对照品溶液引入的相对标准不确定度为:
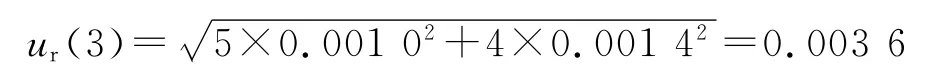
(3)配制含药标准血清时引入的不确定度,ur(4)含药标准血清是由0.1 mL 丙戊酸标准溶液、1 mL空白血清混合而成,其不确定度主要由0.1 mL吹出式分度吸量管和1 mL单标线吸量管引起。
0.1 mLA 级吹出式分度吸量管:
校准:JJG196-2006规定,20 ℃时的容量允差为±0.002mL,按三角分布换算成标准偏差为0.002/=0.000 82 mL。
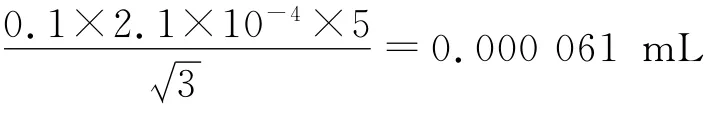
1 mLA 级单标线吸管:
校准:JJG196-2006规定,20 ℃时的容量允差为±0.007 mL。按三角分布换算成标准偏差为0.007/=0.002 8 mL。
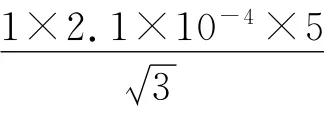
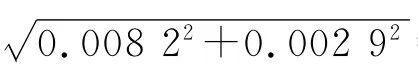
(4)血清样品提取过程,ur(5) 按“2.3”项下方法平行配制低(Low,L)和中(High,H)浓度质控样品各5 份,按“2.3”项下方法处理后进样,进行GC 测定,记录峰面积,按上述回归方程计算浓度值,与配制浓度比较计算回收率,结果见表1。
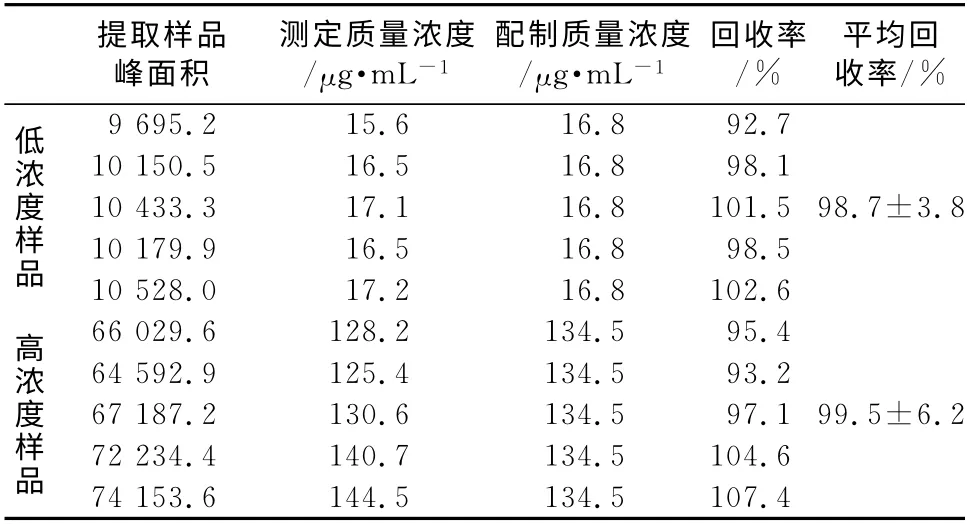
表1 重复性结果回收率结果(n=5)Tab 1 Results of repeatability and recovery(n=5)
回收率的相对标准不确定度:

(5)线性回归过程引入的不确定度ur(6) 生物样本中药物测定时需要先建立标准曲线,然后用标准曲线计算得到未知样品的浓度,直线回归是最常用且最简单的一种,回归方程的建立对计算未知样品的浓度至关重要。按“2.4”项下方法操作,每个浓度测定2次,结果见表2。
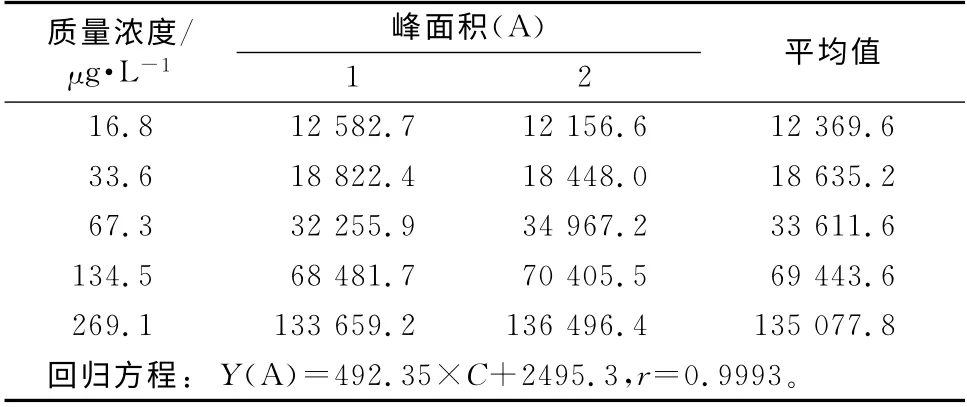
表2 标准曲线结果Tab 2 Results of calibration curve
采用最小二乘法拟合标准曲线计算不确定度。
Yj—第i个标准溶液的第j次峰面积;Xi—第i个标准溶液的浓度;a—斜率;b—截距;P=5(c的测量次数);n=10 标准溶液的测定总次数;a=492.35;b=2 495.3;—重复性样品中丙戊酸的平均浓度,μg·mL-1;=104.26μg·mL-1(标准系列浓度的算术平均值);Ci、Ai—分别为标准系列各浓度对应的丙戊酸浓度和蜂面积。
残余标准偏差为:
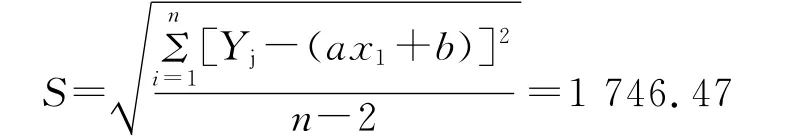
则其相对标准不确定度为:

3.3 合成不确定度的评定 丙戊酸浓度测定的相对标准不确定度为:

丙戊酸浓度测定的标准不确定度为:
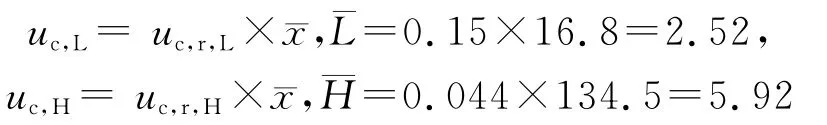
3.4 扩展不确定度的评定 取k=2,此时对应的置信概率为95.45%,丙戊酸浓度测定的扩展不确定度为:UL=k×uc,L=2×2.52=5.04μg·mL-1,UH=k×uc,H=2×5.92=11.84μg·mL-1
人血清中低浓度和中浓度质控样品中丙戊酸的浓度可分别表示为(16.8±5.04)μg·mL-1、(134.5±11.84)μg·mL-1,k=2(95.45%的置信区间)。
4 讨论
固相萃取(SPE)是利用固体吸附剂将液体样品中的目标化合物吸附,与样品的基体和干扰化合物分离,然后再用洗脱液洗脱或加热解吸附,达到分离和富集目标化合物的目的。生物样品中药物具有浓度低、组分复杂、干扰物质多、易受环境影响等特点,采用该方法能提高工作效率、减少试剂污染,可广泛推广。
测量不确定度表征合理地赋予被测量之值的分散性,广义讲是指对测量结果正确性的可疑程度[6]。在生物分析化学领域,测量不确定度的评定越来越受到人们的重视,它与实验室的质量控制(QC)和质量保证(QA)密切相关,可作为方法学验证和水准鉴定的一部分[7]。根据计算出的不确定度分量值,绘制出各不确定度分量的统计直方图(图1),评定显著性不确定度分量,在实际操作中,可以将这些不确定度较大分量的步骤细化,找出影响因素和解决办法,为方法学建立提供科学指导,使测定结果更加准确可靠。图中显示标准曲线的拟合对低浓度样品不确定度的贡献最大,与文献报道相似[12];而血清药物提取(回收率)及仪器测定重复性(精密度)对高浓度样品不确定度的贡献最大。分析其主要原因可能是:(1)血清药物提取时运用固相萃取柱,样品浓度过高不易完全吸附和洗脱,导致产生较大不确定度。(2)气相色谱仪器测定时由于是肉眼观察量取剂量增加不确定性,而且高浓度样品上次进样残留大,因此产生较大不确定度。结果提示,在使用SPE-GC法测定人血中药物浓度时,应加强技术人员的操作水平和技术培训、尽量使用同一规格的量器配制溶液、选择精密的量器和仪器、优化血清前处理方法、选择合适的测定浓度,高浓度样品的测定尤其要注意清洗进样器,同一样品最好同一人员测定等等。
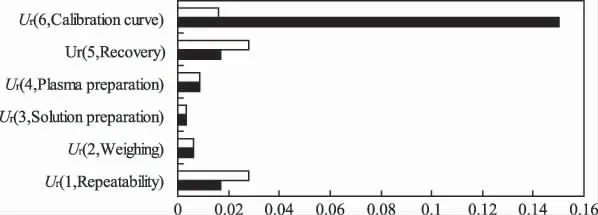
图1 不确定度分量的统计直方图-□-高浓度;-■-低浓度Fig 1 Components of Uncertainty-□-high;-■-low
[1] 张华年,陈渝军,刘智胜.毛细管气相色谱法测定癫痫患儿丙戊酸血药浓度[J].中国医院药学杂志,2005,25(1):46-48.
[2] 张华年,陈渝军,刘智胜.程序升温毛细管气相色谱法监测丙戊酸血药浓度的价值及临床意义[J].儿科药学杂志,2007:14-16.
[3] 周莉华,涂琼,梅步云.HPLC 法测定人血清中丙戊酸钠的浓度[J].中国药房,2011,22(26):2441-2442.
[4] 邹远高,黄英,梁茂植,等.毛细管柱气相色谱法测定血浆中的丙戊酸浓度[J].华西药学杂志,2002,17(1):51-52.
[5] 吴妙莲,黄云,张慧芬.273例癫痫患儿血清丙戊酸浓度和剂量分析[J].中国医院药学杂志,2011,31(17):1444-1447.
[6] 国家质量技术监督局.测量不确定度评定与表示[J].JJF1059-1999.
[7] Burns M.Current practice in the assessment and control of measure-ment uncertainty in bio-analytical chemistry[J].Trac Trend Anal Chem,2004,23(5):393.
[8] 国家质量监督检验检疫总局.常用玻璃量器检定规程[S].JJG196-2006.
[9] 郭兰典.商品检测不确定度评定实例[M].北京.中国计量出版社,2004:1-5.
[10]中国实验室国家认可委员会.化学分析中不确定度的评估指南[M].北京.中国计量出版社,2002:103-117.
[11]粟晓黎,李冠民,金少鸿.药品检验一般检测项目不确定度评定研究-1.B类评定[J].药物分析杂志,2005,25(6):699.
[12]严蓓,杨晨,李扬.HPLC 法测定人血浆中奥硝唑浓度的不确定度评定[J].药物分析杂志,2011,31(9):1797-1803.
猜你喜欢
中国民间疗法(2021年10期)2021-07-22
中国现代医药杂志(2020年10期)2020-12-14
天津医科大学学报(2019年3期)2019-08-13
山东化工(2018年15期)2018-09-20
中国卫生标准管理(2015年5期)2016-01-14
首都食品与医药(2015年18期)2015-11-03
中国学术期刊文摘(2015年8期)2015-10-29
四川生理科学杂志(2014年2期)2014-02-28
现代检验医学杂志(2014年1期)2014-02-06
长江大学学报(自科版)(2013年33期)2013-03-11
